




【美国进口清关】FDA 医疗器械监管与器械分类

 5396
5396

一键完成百千笔付款,超低费率+极速到账,一年轻松帮你省下数十万。
美国食品药品监督管理局 FDA 根据医疗器械的风险程度以及为合理确保其安全性和有效性所需的监管措施(Regulatory controls),将其分为三类:I 类、II 类和 III 类。I 类器械通常对患者和/或使用者构成的风险最低,而 III 类器械构成的风险最高。
医疗器械监管概述
美国食品药品监督管理局 FDA 的医疗器械和放射健康中心 CDRH(Center for Devices and Radiological Health)负责监管生产(Manufacture)、重新包装(Repackage)、重新贴标(Relabel)和/或进口在美国销售的医疗器械的企业(Firms)。此外,CDRH 还监管辐射电子产品(Radiation-emitting electronic products)(医用和非医用),例如激光器(Lasers)、X 射线系统(X-ray systems)、超声波设备(Ultrasound equipment)、微波炉(Microwave ovens)和彩色电视机(Color televisions)。
医疗器械分为 I 类、II 类和 III 类。从 I 类到 III 类,监管要求依次递增。医疗器械分类法规(Device classification regulation)规定了各类器械的监管要求。大多数 I 类医疗器械(Class I devices)器械可豁免于(Exempt)上市前通知(Premarket Notification)510(k);大多数 II 类医疗器械(Class II devices)器械需要(Require)提交上市前通知(Premarket Notification)510(k);大多数 III 类医疗器械(Class III devices)器械需要(Require)提交上市前批准 PMA(Premarket Approval)。
在美国销售的医疗器械制造商(Manufacturers)必须遵守的基本监管要求(Basic regulatory requirements)包括:
机构注册(Establishment registration) 器械列名(Medical Device Listing) 上市前通知 510(k)(除非获得豁免),或上市前批准 PMA(Premarket Approval) 研究用医疗器械豁免 IDE(Investigational Device Exemption) 质量体系 OS(Quality System)法规 标签要求(Labeling requirements);以及 医疗器械报告 MDR(Medical Device Reporting)
机构注册(Establishment registration) - 21 CFR Part 807
医疗器械制造商(包括美国国内外制造商)和初始分销商(进口商)必须向 FDA 注册其机构(Establishments)。所有机构注册(Establishment registrations)均须以电子方式提交,除非获得 FDA 豁免。所有注册信息必须在每年 10 月 1 日至 12 月 31 日期间进行年度验证(Verified annually)。除注册外,外国制造商(Foreign manufacturers)还必须指定一名美国代理(U.S. Agent)。自 2007 年 10 月 1 日起,大多数机构均须缴纳机构注册费(Establishment registration fee)。
器械列名(Medical Device Listing) - 21CFR Part 807
制造商(Manufacturers)必须向 FDA 列名(List)其医疗器械。需要进行医疗器械列名的机构(Establishments)包括:
制造商(Manufacturers) 合同制造商(Contract manufacturers) 合同灭菌服务商(Contract sterilizers) 再包装商和再贴标商(Repackagers and relabelers) 产品规格制定方(Specification developers) 一次性医疗器械再处理商(Reprocessors single-use devices) 再制造商(Remanufacturer) 直接向最终用户销售配件和组件的制造商(Manufacturers of accessories and components sold directly to the end user) "仅限出口 "器械的美国制造商(U.S. manufacturers of "export only" devices)
上市前通知(Premarket Notification)510(k) - 21 CFR Part 807 Subpart E
如果你的医疗器械需要提交上市前通知 510(k),则必须收到 FDA 授权的实质等同认定函(A letter of substantial equivalence)后该器械才能上市销售(Commercially distribute)。510(k) 必须证明(Demonstrate)该器械与以下已在美国合法上市的医疗器械实质等同 SE(Substantially equivalent):(1) 1976 年 5 月 28 日之前上市的医疗器械,或 (2) 经 FDA 认定为实质等同的医疗器械。
2002 年 10 月 26 日,《2002 年医疗器械用户费用和现代化法案》(Medical Device User Fee and Modernization Act of 2002)正式生效。该法案授权 FDA 对医疗器械上市前通知 510(k) 审查收取费用。申请费(Application fee)适用于传统(Traditional)、简化(Abbreviated)和特殊(Special)510(k)。
大多数 I 类器械(Class I devices)和部分 II 类器械(Class II devices)无需提交上市前通知 510(k)。豁免器械列表(A list of exempt devices)可在以下 CDRH 公共数据库查询:
510(k) 器械豁免数据库(510(k) Exempt Devices):http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpcd/315.cfm
如果你计划向 FDA 提交 I 类或 II 类器械的 510(k) 申请(510(k) application),由经认可的机构(Accredited Persons)进行 510(k) 审查可能会对你有所帮助。FDA 已认可 12 家机构对 670 种器械进行初步审查(Primary review)。根据法律规定,FDA 必须在收到经认可机构的建议后 30 天内发布最终决定(Final determination)。
上市前批准 PMA(Premarket Approval) - 21 CFR Part 814
需要获得 PMA 批准的产品属于 III 类医疗器械(Class III devices),即存在重大疾病或损伤风险的高风险器械,或通过 510(k) 流程被认定不与 I 类和 II 类器械实质等同的医疗器械。上市前批准 PMA 流程更为复杂,包括提交临床数据以支持针对该器械的声明。
自 2003 财年(2002 年 10 月 1 日至 2003 年 9 月 30 日)起,医疗器械用户费用适用于原始 PMA(产品授权许可)及某些类型的 PMA 补充协议(Supplements)。
研究用医疗器械豁免 IDE(Investigational Device Exemption) - 21CFR Part 812
研究用医疗器械豁免 IDE 允许将研究用器械用于临床研究(Clinical study),以收集向 FDA 提交上市前批准 PMA 申请或上市前通知 510(k) 所需的安全性和有效性数据。对于具有重大风险的器械,其临床研究必须获得 FDA 和机构审查委员会 IRB(Institutional Review Board) 的批准后才能开始。对于低风险的器械,其临床研究只需获得 IRB 的批准后即可开始。
质量体系 QS 法规(Quality System Regulation) - 21 CFR Part 820
质量体系法规涵盖医疗器械设计(Designing)、采购(Purchasing)、制造(Manufacturing)、包装(Packaging)、标签(Labeling)、储存(Storing)、安装(Installing)和维护(Servicing)过程中所使用的方法、设施和控制措施的相关要求。生产设施(Manufacturing facilities)需接受 FDA 检查,以确保符合质量体系要求。
标签(Labeling) - 21 CFR Part 801
标签包括医疗器械上的标签以及器械附带的描述性和信息性文献(Descriptive and informational literature)。
医疗器械报告 MDR(Medical Device Reporting) - 21 CFR Part 803
根据医疗器械报告计划,任何可能导致或促成死亡或严重伤害的器械事故都必须向 FDA 报告。此外,某些故障(Malfunctions)也必须报告。MDR 法规是 FDA 和制造商识别和监控涉及医疗器械的重大不良事件的机制。该法规(Regulation)的目标是及时发现并纠正问题。
美国医疗器械监管历史
美国食品药品监管管理局 FDA 是美国历史最悠久的综合性消费者保护机构(Comprehensive consumer protection agency)。FDA 对食品和药品的监管始于 1906 年,当时美国时任总统西奥多·罗斯福(Theodore Roosevelt)签署了《纯净食品药品法案》(Pure Food and Drugs Act)。
20 世纪 60 年代和 70 年代,为了响应公众对医疗器械加强监管的诉求,美国国会通过了《联邦食品、药品和化妆品法案》医疗器械修正案(Medical Device Amendments to the Federal Food, Drug, and Cosmetic Act)。1982 年,FDA下属负责监管医疗器械和放射产品的部门合并,成立了医疗器械和放射健康中心 CDRH(Center for Devices and Radiological Health)。
以下时间年表概述了美国医疗器械立法史上的里程碑事件:
1906 年:《纯净食品药品法案》(Pure Food and Drugs Act)(有时也称为《联邦食品药品法案》)(Federal Food and Drugs Act)
建立了如今 FDA 的前身 禁止在州际间进行掺假(Adulterated)和误标(Misbranded)的食品及药品的商业贸易活动
1938 年:《联邦食品、药品和化妆品法案》(FD&C Act)(Federal Food, Drug, and Cosmetic Act)
授权 FDA 监管和监督(Regulation and oversight)医疗产品的主要法规 将州际贸易禁令的范围扩大至误标和掺假的化妆品和治疗性医疗器械 工厂检查权(Authority for factory inspections)
1944 年:《公共卫生服务法案》(Public Health Service Act)
建立实验室认证体系 扩大对生物制品(Biologics)的监管
1968 年:《辐射控制健康与安全法案》(Radiation Control for Health and Safety Act)
旨在最大限度地减少电子产品辐射和强磁场照射 制定了辐射发射产品(Radiation-emitting products)的性能标准,如诊断用 X 光机(Diagnostic x-ray machines)、核磁共振成像仪(MRIs)、微波设备(Microwave)、超声或透热设备(Ultrasound or diathermy devices)、紫外线设备(UV devices)和激光设备(Laser devices)等
1970 年:尼克松成立库珀委员会(Cooper Committee)
由西奥多·库珀(Theodore Cooper)医学博士(时任美国国家心肺研究所所长)担任主席,研究医疗器械立法的必要性 建议任何新的立法都应专门针对医疗器械(Devices),因为医疗器械带来的问题不同于药物(Drugs) 引入了基于风险的医疗器械分类概念
1976 年:《联邦食品、药品和化妆品法案》医疗器械修正案(Medical Device Amendments to the FD&C Act)
旨在为医疗器械的安全性和有效性提供合理保证 为所有医疗器械创建了基于风险的三级分类系统 建立了新型医疗器械(1976 年 5 月 28 日之前未上市或经过重大修改的医疗器械)进入市场的监管途径:上市前批准(Premarket Approval)PMA 和上市前通知(Premarket notification)510(k) 为在患者中进行研究的新型研究用医疗器械创建了监管途径(研究用医疗器械豁免 IDE)(Investigational Device Exemption) 建立了几项关键的上市后要求(Postmarket requirements):向 FDA 注册机构和器械列名、良好生产规范 GMPs 以及涉及医疗器械的不良事件报告 授权 FDA 禁用器械
1977 年:医疗器械和诊断产品局(Bureau of Medical Devices and Diagnostic Products)更名为医疗器械局(Bureau of Medical Devices)
1990 年:《安全医疗器械法案》SMDA(Safe Medical Devices Act)
通过以下措施改进医疗器械上市后的监管(Postmarket surveillance):要求医院和疗养院等用户设施(User facilities)报告涉及医疗器械的不良事件;如果器械故障可能导致永久性伤害或死亡,授权 FDA 要求制造商对永久植入式医疗器械进行上市后检测(Postmarket surveillance) 授权 FDA 下令召回医疗器械,并对违反《联邦食品、药品和化妆品法案》(FD&C Act)的行为处以民事处罚(Civil penalties) 明确了实质等同定义(Substantial equivalence)(通过 510(k) 计划销售医疗器械的标准) 修改了制定(Establishment)、修订(Amendment)或撤销(Revocation)性能标准的程序 设立了人道主义用途医疗器械 HUD(Humanitarian Use Device)/ 人道主义器械豁免计划 HDE(Humanitarian Device Exemption),以鼓励开发针对罕见疾病(Rare diseases)的器械
1992 年:《乳腺 X 光检查质量标准法案》MQSA(Mammography Quality Standards Act)
要求乳腺 X 光检查设施必须获得认可和联邦认证并符合质量标准。初次认证后,设施必须通过联邦或州检查员的年度检查
1997 年:《FDA 现代化法案》FDAMA(Food and Drug Administration Modernization Act)
制定了“最少负担”(Least burdensome)原则的上市前审查规定 允许经认可的第三方机构对某些器械进行初步上市前审查(Initial premarket reviews) 允许在新版本器械的上市前申请中使用早期版本器械研究的数据 为扩大研究用医疗器械(Investigational devices)的使用范围提供了便利 建立了 De Novo 项目,通过该项目,新型低风险至中等风险器械可以被归类为 I 类或 II 类,而不是自动归类为 III 类
2002 年:《医疗器械用户费用和现代化法案》MDUFMA(Medical Device User Fee and Modernization Act)
授权 FDA 对特定医疗器械上市前申请(Premarket submissions)收取用户费用,以帮助 FDA 提高医疗器械上市申请审查的效率、质量和可预测性 制定了小型企业认定计划 SBD(Small Business Determination),允许降低符合条件的小型企业医疗器械上市前申报的审批费用 为 FDA 某些上市前申报的决策制定了绩效目标(Performance goals) 针对“再处理”(Reprocessed)器械制定了新的监管要求 授权医疗器械企业进行电子注册 设立了组合产品办公室(Office of Combination Products)
2007 年:《FDA 修正法案》FDAAA(Food and Drug Administration Amendments Act)
重新授权医疗器械用户费用计划(MDUFA II),包括改进上市前审查时间 要求所有注册和列名(Registration and listing)均以电子方式进行 要求 FDA 为医疗器械建立唯一标识 UDI(Unique device identification)系统,要求器械标签带有唯一标识符
2012 年:《FDA 安全与创新法案》FDASIA(Food and Drug Administration Safety and Innovation Act)
重新授权医疗器械用户费用计划(MDUFA III),包括改进上市前审查时间并增加与行业共享的结果目标 创建了直接的 De Novo 途径(Pathway),允许将新型、低风险至中等风险的医疗器械归类为 I 类或 II 类(而不是 III 类),而无需先提交 510(k) 修改了与不批准 IDE 相关的标准 允许 FDA 与外国政府合作,协调监管要求 要求 FDA 在提交申请的持有人(Holder)提出重大决策要求时提供实质性摘要(Substantive Summary) 扩大了 “最少负担 ”(Least burdensome)原则在上市前审查中(Premarket reviews)的应用范围
2016 年:《21 世纪治愈法案》(21st Century Cures Act)
授权制定或修订旨在加快患者获得新医疗器械的政策和程序,包括:
将 FDA 对突破性医疗器械的快速审查程序编入法律 扩大 “最少负担 ”(Least burdensome)原则在上市前审查中(Premarket reviews)的应用范围 简化医疗器械上市前通知(510(k))豁免流程 将美国人道主义用途医疗器械 HUD(Humanitarian Use Device)认定所需的患者人数估计值从每年“少于4,000人”提高到“不超过8,000人” 允许使用中央机构审查委员会 IRB(Institutional Review Board)监督,而不是只要求地方机构审查委员会监督 IDE 和 HDE 活动 授权 FDA 修订对组合产品的监管规定 将提交标准认可/不认可申请的流程纳入法律
通过定义可以和不可以作为医疗器械进行监管的医疗软件类别,阐明了如何监管某些数字健康产品(Digital health products)/数字医疗产品。
2017 年:《FDA 再授权法案》FDARA(Food and Drug Administration Reauthorization Act)
重新授权医疗器械用户费用计划(MDUFA IV),包括缩短上市前审查时间,以及对国家卫生技术评估系统 NEST(National Evaluation System for health Technology)和患者意见征询等战略举措的投资 授权对医疗器械生产机构(Device establishments)进行基于风险的检查安排,并规定与医疗器械生产机构检查相关的其他流程改进措施 将配件分类(Accessory classification)与母体器械(Parent device)分类分离 要求 FDA 进行至少一个试点项目,以探索真实证据如何改善上市后监管(Postmarket surveillance)
2020 年:《新冠病毒援助、救济和经济安全法案》CARES Act(Coronavirus Aid, Relief, and Economic Security Act)
要求某些医疗器械制造商在突发公共卫生事件期间或之前向 FDA 提交生产中断或永久停止的通知,以增强 FDA 识别、预防和缓解医疗产品供应链可能出现短缺的能力 明确规定 FDA 可以为在外国医疗器械机构(Foreign device establishment)生产并运往其他国家的医疗器械颁发认证
2022 年:《FDA 用户费用重新授权法案》FDAUFRA(FDA User Fee Reauthorization Act of 2022)
重新授权医疗器械用户费用计划(MDUFA V),包括对上市前活动(如预提交)的流程改进和绩效目标,以及对战略举措(如患者科学和参与以及国际协调工作)的投资 在 MDUFA V 的后期,根据绩效目标达成情况和招聘基准(Hiring benchmarks),制定了新的费用调整方案 为 MDUFA 结转余额设定 13 周的上限,并单独资助了一项新的全产品生命周期 TPLC(Total Product Life Cycle)咨询计划 TAP 试点项目
2022 年:《2022 年食品药品综合改革法案》FDORA(Food and Drug Omnibus Reform Act of 2022)
加强对医疗器械机构(Device establishments)的监督,包括授权进行远程监管审计和检查进行器械研究的设施的新权力 明确授权 FDA 批准或许可(Approve or clear)具有预定变更控制计划 PCCP(Predetermined change control plan)的医疗器械 要求在某些器械(称为网络设备)(Cyber devices)的上市前申请(Premarket submissions)中提供有关网络安全的信息,并要求此类器械的申报方(Sponsors)必须确保其网络安全 明确规定 FDA 可以禁止一种或多种预期用途的医疗器械,并且被禁器械不属于合法上市的器械 要求某些器械临床研究的发起人提交一份多元化行动计划,其中包含招募目标,并计划在 FDA 发布该主题的最终指南后 180 天内开始实现这些目标 允许对在外国医疗器械机构(Foreign device establishment)生产并运往其他国家的医疗器械进行认证,前提是该器械也在美国销售,且满足其他标准 从 2025 财年开始,为面临财务困难的小型企业提供新的注册费减免政策
2022 年:《预防和应对现有病毒、新威胁和流行病法案》(预防流行病法案)(Preparing for and Responding to Existing Viruses, Emerging New Threats, and Pandemics Act)(PREVENT Pandemics Act)
对假冒伪劣医疗器械处以更严厉的处罚 明确规定 FDA 可在公共卫生紧急事件之外接收自愿的器械短缺通知,并指示该机构创建一份须强制通知的医疗器械类型清单 明确规定 FDA 可依赖合格的第三方审查体外诊断器械(Vitro diagnostic devices)的紧急使用授权 EUA(Emergency use authorizations)申请
FDA 医疗器械分类
美国食品药品监督管理局 FDA 已为大约 1700 种不同的通用类别医疗器械(Generic types of devices)制定了分类,并将它们归入 16 个医学专科领域(Medical specialties),称为 “医疗器械分类专业组”(Panels)。根据确保医疗器械安全性和有效性所需的监管程度,每种通用类别医疗器械(Generic types of devices)都被归入三个监管类别之一。这三个类别及其适用的要求如下:
医疗器械类别和监管控制(Device Class and Regulatory Controls):
1,I 类一般管制要求(Class I General Controls)
有豁免(With Exemptions) 无豁免(Without Exemptions)
2,II 类一般管制要求和特殊管制要求(Class II General Controls and Special Controls)
有豁免(With Exemptions) 无豁免(Without Exemptions)
3,III 类一般管制要求和上市前批准(Class III General Controls and Premarket Approval)
除其他事项外,器械所属的类别(Class)决定了 FDA 批准上市所需的上市前申报(Premarketing submission/application required)类型等。如果器械被归类为 I 类或 II 类,并且未获豁免(Not exempt),则必须提交 510k 以获得上市许可。所有被归类为豁免的器械均受豁免限制的约束。器械豁免限制在 21 CFR xxx.9 中规定,其中 xxx 指的是第 862-892 部分(Parts 862-892)。对于 III 类器械,除非该器械属于修正案前医疗器械(在 1976 年医疗器械修正案通过之前已上市,或与此类器械实质等同 SE),且未要求提交 PMA,否则将需要提交上市前批准申请 PMA(Premarket approval application)。在这种情况下,510k 申报将是上市途径(Route to market)。
器械分类取决于器械的预期用途(Intended use)以及适应症(Indications for use)。例如,手术刀的预期用途是切割组织。当器械标签中添加更具体的适应症(例如“用于在角膜上切开”)时,则构成预期用途的一个子集。
此外,医疗器械分类(Classification)是基于风险的,也就是说,器械(Device)对患者和/或用户造成的风险是决定其分类类别的主要因素。 I 类医疗器械风险最低, III 类医疗器械风险最高。
如上所述,所有类别的器械均受一般管制要求(General Controls)的约束。一般管制要求是《食品、药品和化妆品法案》(FD&C Act)的基本要求,适用于所有 I、II 和 III 类医疗器械。
要查找器械的分类以及是否存在任何豁免,需要找到作为你器械分类法规的法规编号(Regulation number)。
有两种方法可以实现此目的:直接访问分类数据库(Classification database)并搜索器械名称的一部分,或者,如果你知道器械所属的医疗器械分类专业组(Device panel )(医学专科(领域))(Medical specialty),则直接进入该分类的列表并识别你的器械和相应的法规。
如果你已经知道相应的专业组(Panel),你可以直接访问 CFR,通过阅读分类器械列表(List of classified devices)来查找器械的分类;如果不确定,可以使用产品代码分类数据库中的关键字目录。在大多数情况下,该数据库将确定 CFR 中的分类法规。你也可以查看下面的分类法规(Classification regulations),了解各种产品的信息以及 CDRH 如何对其进行监管。
CFR 862:https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-862
CFR 中的每个医疗器械分类专业组(Classification panel)都以该专业组中分类的器械列表开始。每个分类器械都有一个与之关联的 7 位数编号,例如 21 CFR 880.2910 临床电子体温计(Clinical electronic thermometer)。
部分 I 类医疗器械(Class I devices)可豁免于上市前通知(Premarket notification)和/或部分良好生产规范 GMPs(Good manufacturing practices regulations)的规定。大约有 572 种(占 I 类器械的 74%)的 I 类医疗器械(Class I devices)可免于上市前通知流程(Premarket notification process)。
这些豁免信息列于 21 CFR 的分类法规(Classification regulations)中,并已汇总于《医疗器械豁免》(Medical Device Exemptions)数据库中。
510(k) 和 GMP 要求豁免器械数据库:https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpcd/315.cfm
如何确定你的产品是否属于医疗器械
医疗器械介绍
根据 FDA 监管法规的定义,医疗器械(Medical devices)的范围从简单的压舌板和便盆到复杂的可编程心脏起搏器和闭环人工胰腺系统,种类繁多。此外,医疗器械还包括体外诊断 IVD 产品,例如试剂、检测试剂盒和血糖仪。某些具有医疗用途或声称具有医疗用途的发射辐射的电子产品(Radiation-emitting electronic products)也被视为医疗器械。这些产品包括超声诊断产品、X 光机和医疗激光器。
医疗器械确定步骤
以下步骤可能有助于确定产品是否作为医疗器械受 FDA 监管。
步骤一:
确定产品是否符合《联邦食品、药品和化妆品法案》第 201(h) 条(Section 201(h) of the Food, Drug & Cosmetic Act)中规定的医疗器械定义。 步骤二:
确定产品是否存在合适的分类。
步骤一:确定产品是否符合医疗器械的定义
如果产品符合《联邦食品、药品和化妆品法案》第 201(h) 条(Section 201(h) of the Food, Drug, and Cosmetic Act)对医疗器械的定义,则 FDA 就会将该产品视为医疗器械,并对其进行监管。
根据《联邦食品、药品和化妆品法案》第 201(h) 条(Section 201(h) of the FD&C Act)的规定,医疗器械(Device)是指:
仪器(Instrument)、设备(Apparatus)、器具(Implement)、机器(Machine)、装置(Contrivance,)、植入物(Implant)、体外试剂(In vitro reagent)或其他类似或相关物品,包括以下组件、零件或附件:
(A)在美国国家处方集 NF(National Formulary)、美国药典 USP(United States Pharmacopoeia)或其任何补充中得到认可, (B)用于诊断人类或其他动物的疾病或其他病症,或用于治愈、缓解、治疗或预防疾病,或 (C)旨在影响人类或其他动物的身体结构或任何功能,并且不通过人类或其他动物体内或身体上的化学作用实现其主要预期目的,也不依赖于代谢来实现其主要预期目的。术语“器械”(Device)不包括根据第 520(o) 条(Section 520(o))排除的软件功能。
2016 年 12 月,《21 世纪治愈法案》(21st Century Cures Act)第 3060(a) 条(Section 3060(a))对《联邦食品、药品和化妆品法案》FD&C Act(Federal Food, Drug, and Cosmetic Act)第 520(o) 条(Section 520(o))进行了修订,将某些软件功能(Certain software functions)(Software functions)(包括旨在维持或鼓励健康生活方式,但与疾病或病症的诊断、治愈、缓解、预防或治疗无关的功能)从《FD&C法案》第201(h)条(Section 201(h) of the FD&C Act)的医疗器械定义中删除。例如,用于维持或促进健康生活方式的软件功能,与疾病或病症(Disease or condition)的诊断、治疗、缓解、预防或护理(Diagnosis, cure, mitigation, prevention, or treatment)无关;用作电子病历的软件功能;用于传输、存储、格式转换或显示临床实验室检测或其他设备数据、结果或发现但其目的并非对这些数据进行解释或分析的软件功能。
要确定产品是否符合医疗器械的定义,应先确定产品的预期用途(Intended use)和适应症(Indications for use)。一旦明确了产品的预期用途和适应症后,就可以确定产品是否符合医疗器械的定义。
预期用途(Intended use):
该医疗器械的一般用途(General purpose)或其功能(Function)。医疗器械的预期用途包括其适应症。 适应症(Indications for use):
对该医疗器械将用于诊断(Diagnose)、治疗(Treat)、预防(Prevent)、治愈(Cure)或缓解(Mitigate)的疾病(Disease)或病症(Condition)的总体描述(General description),包括对该医疗器械适用的患者群体的描述(21 CFR 814.20(b)(3)(i))。
如果产品符合《联邦食品、药品和化妆品法案》(FD&C Act)第 201(h) 条(Section 201(h))中的定义,则该产品将作为医疗器械受到 FDA 的监管,并将受到上市前和上市后监管控制(Subject to premarket and postmarket regulatory controls)。
步骤二:确定产品是否存在合适的分类
在确定产品是否作为医疗器械收到监管时,可以搜索可能适用于你的产品的现有产品分类(Product classifications)。找到一个能够描述你产品预期用途或设计(Design)的现有分类,是判断其是否属于医疗器械(Medical device)的一个重要指标。
以下概述了三种确定你的产品是否存在相应产品分类(Product classification)的方法。
方法 1:搜索产品分类数据库(Product Classification Database)
可以搜索 FDA 产品分类数据库(FDA Product Classification Database)来确定是否存在适用于你的产品的现有产品分类:
FDA 产品分类数据库(FDA Product Classification Database):https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm
使用快速搜索(Quick Search)功能按关键词进行搜索。请注意,你可能需要使用描述你产品的多个关键词进行多次搜索(例如,同时搜索“stent”和“stents”)。 使用高级搜索(Advanced Search)功能按产品代码(Product code)、法规编号(Regulation number)或器械类别(Device class)进行搜索。
方法 2:搜索类似器械(Similar Devices)
如果你发现在美国市场上存在已合法销售的类似医疗器械,你可以查找允许其上市许可(Permits marketing authorization)的 FDA 信函或命令(FDA letter or order)。该函件或命令中关于类似器械类型的信息可能有助于你确定自身医疗器械的分类。
FDA 批准上市许可(Permit marketing authorization)的决定是公开信息,可以通过搜索以下 CDRH 公共数据库使用“快速搜索”(Quick Search)或“高级搜索”(Advanced Search)功能来查询:
上市前批准 PMA 数据库:
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMA/pma.cfm 上市前通知 510(k) 数据库:
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/pmn.cfm De Novo 新分类申请数据库:
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/denovo.cfm 人道主义医疗器械豁免 HDE 数据库(Humanitarian Device Exemption):
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfhde/hde.cfm
上市前批准 PMA(Premarket Approval):
大多数 III 类(高风险)(Class III)医疗器械在合法上市(Legally marketed)前都需要获得上市前批准 PMA(Premarket Approval)申请的批准。该数据库包含已获上市前批准的医疗器械,并包含批准令(Approval order),安全性和有效性摘要(Summary of Safety and Effectiveness)以及获批器械的标签(原始 PMA 和专家委员会审评型 PMA 补充申请)(Original PMAs and panel-track supplements) 上市前通知(Premarket Notification)510(k):
大多数 II 类(中等风险)(Class III)医疗器械在合法上市前都需要获得 FDA 的 510(k) 许可(510(k) clearance)。 De Novo:
De Novo 提供了一种对低风险至中等风险的新型器械(Novel devices)进行分类的可能途径。该数据库包含 De Novo 医疗器械分类令(Classification orders)和透明性摘要(Transparency summaries)。 人道主义器械豁免 HDE(Humanitarian Device Exemption):
HDE 为帮助罕见疾病患者的医疗器械上市提供了一种可行的途径。该数据库包含已获 HDE 批准的器械,并包含批准令(Approval order),安全性和预期效益摘要(Summary of Safety and Probable Benefit)以及经批准的人道主义医疗器械标签(Labeling for the approved device)。
注意:大多数 I 类(Class I)医疗器械和部分 II 类医疗器械(Class II)可能未列于上述数据库中,因为它们属于豁免器械(Exempt),在上市前不需要 FDA 的审查(FDA's review)。
方法 3:通过器械列名搜索类似器械(Search for Similar Devices by Device Listing)
可以通过查看合法上市医疗器械的器械列名信息来查找其产品分类。医疗器械列名信息可通过 FDA 的机构注册&器械列名(Establishment Registration and Device Listing)数据库查询可以通过使用快速搜索或高级搜索功能(Quick or Advanced Search feature),来查找器械的列名信息(Device listing)。
如果你的产品不符合医疗器械的定义,则可能由 FDA 下属的其他中心(Center)监管:
生物制品评审与研究中心 CBER(Center for Biologics Evaluation and Research)
:负责监管生物制品(Biological products)。 药品评审与研究中心 CDER(Center for Drug Evaluation and Research)
:负责监管人类药物(Human drugs)。如果产品的主要预期用途是通过化学作用或经人体代谢来实现的,则该产品可被视为药物(Drug)。 兽医用药与产品中心 CVM(Center for Veterinary Medicine)
:负责监管拟用于动物的产品(如动物药、饲料、宠物食品等)。 烟草制品中心 CTP(Center for Tobacco)
:负责监管烟草制品(Tobacco products)。
医疗器械分类专业组
什么是医疗器械分类专业组(Device Classification Panels)
大多数医疗器械(Medical devices)可以通过在《联邦法规汇编》 CFR(Code of Federal Regulations)第 21 编(Title 21)第 862-892 部分(Parts 862-892)中找到相应的描述并进行分类。
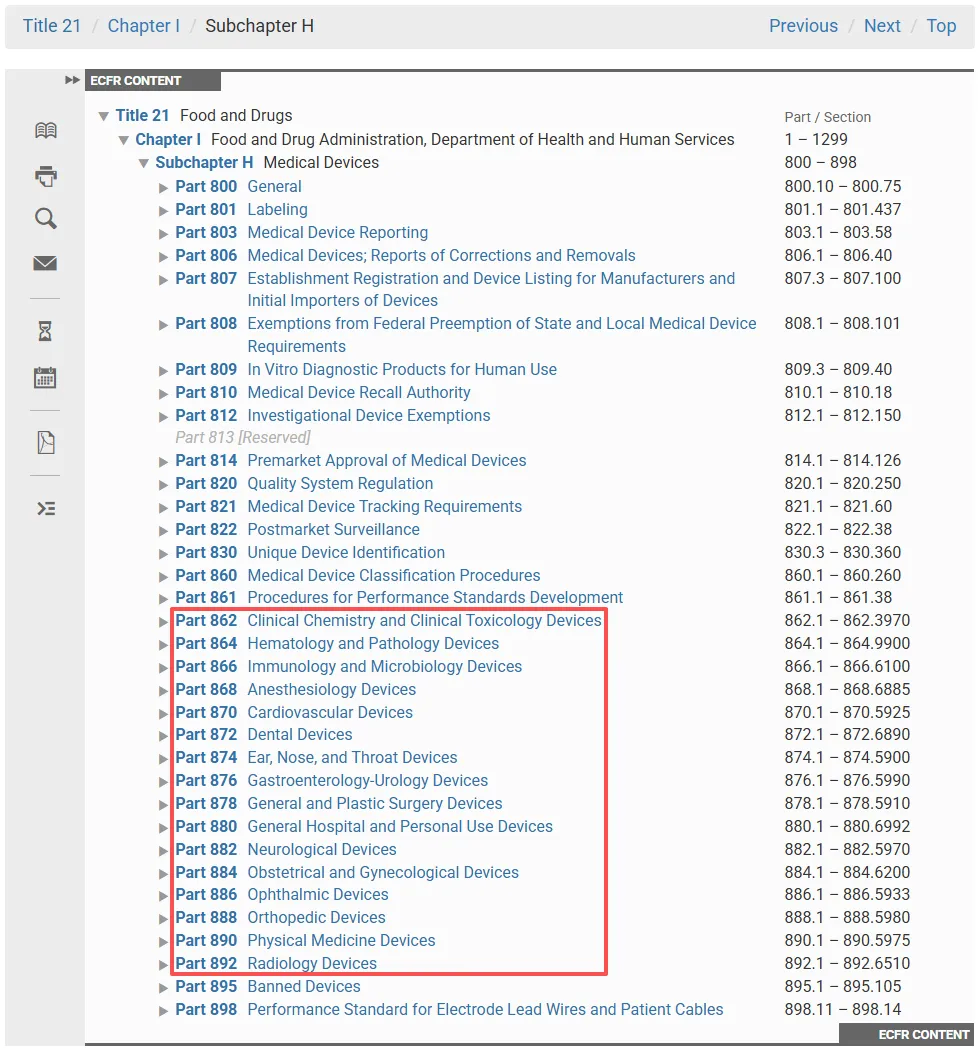
美国食品药品监管管理局 FDA 已对 1700 多种不同类型的医疗器械进行了分类和描述,并在 CFR 中将它们划分为 16 个医学专业领域(Medical specialty)“专业组”(Panels),例如心血管器械(Cardiovascular devices)或耳鼻喉器械(Ear, Nose, and Throat devices)。这些医疗器械分类专业组(Panels)位于 CFR 第 862 至 892 部分。
对于 FDA 分类的每种器械,CFR 都提供了一般描述(General description)、器械所属的类别(即 I 类、II 类或 III 类)以及有关上市要求的信息(Marketing requirements)。你的器械应符合 21 CFR 第 862-892 部分中的分类法规(Classification regulation)定义。
如何查找分类法规(Classification Regulations)
重新分类(Reclassification)
美国食品药品监督管理局 FDA 通常根据医疗器械相关的风险程度,以及评估能够合理保证器械安全性和有效性的监管力度,对医疗器械进行分类。医疗器械分为三类:I 类、II 类和 III 类。根据《联邦食品、药品和化妆品法案》第 513(a) 条(Section 513(a) of the Federal Food, Drug and Cosmetic Act)(21 U.S.C. 360c(a))及其执行条例 21 CFR 860.3 的规定,某类医疗器械的监管类别(Regulatory class)可通过重新分类流程(Process of reclassification)进行变更。重新分类的主要目的是根据有关医疗器械类型安全性和有效性的最新信息,实施相应的监管控制级别(Appropriate level of regulatory controls)。重新分类针对的是医疗器械类型(Device type),而非单个医疗器械(Individual device)。
《联邦食品、药品和化妆品法案》(FD&C Act)中有关医疗器械重新分类的条款如下:
Section 513(e):
适用于已分类的医疗器械(Classified devices),但目前根据第 513(f)(1) 条(Section 513(f)(1))分类为 III 类的医疗器械除外。 Section 513(f)(3):
适用于根据第 513(f)(1) 条(Section 513(f)(1))自动分类为 III 类的修正案后医疗器械。
注:在 1976 年 5 月 28 日《联邦食品、药品和化妆品法案》医疗器械修正案(Medical Device Amendments to the FD&C Act)颁布生效之前尚未上市(Commercial distribution)的医疗器械(通常称为修正案后医疗器械)(Postamendments devices),根据《联邦食品、药品和化妆品法案》第 513(f)(1) 条(Section 513(f)(1) of the FD&C Act)均自动归类为 III 类医疗器械,无需 FDA 制定任何规则程序(Rulemaking process),也无论风险程度如何。除非且直至该器械通过该法案第 513(f)(2) 条(513(f)(2) of the FD&C Act)规定的新分类程序(De Novo process)进行分类、重新归类为 I 类或 II 类医疗器械,或者 FDA 根据《联邦食品、药品和化妆品法案》第 513(i) 条(Section 513(i) of the FD&C Act)发布命令,认定该器械与无需上市前批准 PMA 的对比器械(Predicate device)实质等同(Substantially equivalent),否则这些器械仍属于 III 类,需要获得上市前批准 PMA。
《联邦食品、药品和化妆品法案》第 513(e) 条(Section 513(e) of the FD&C Act)
适用于对已分类医疗器械的重新分类,但目前根据第 513(f)(1) 条(Section 513(f)(1))分类为 III 类的医疗器械除外。
2012 年 7 月 9 日,《FDA 安全与创新法案》FDASIA(Food and Drug Administration Safety and Innovation Act)正式生效,将《联邦食品、药品和化妆品法案》第 513(e) 条(Section 513(e) of the FD&C Act)规定的重新分类程序(Reclassification process)从制定规则(Rulemaking)改为行政命令程序(Administrative order process)。
对于已分类的医疗器械(但目前根据第 513(f)(1) 条分类为 III 类的医疗器械除外),美国食品药品监督管理局 FDA 可主动启动器械类型的重新分类程序,或响应相关方提出的重新分类申请。无论哪种情况,重新分类(Reclassification)的依据均在于该医疗器械的新信息。根据《联邦食品、药品和化妆品法案》第 513(e) 条对医疗器械进行重新分类,FDA 在最终确定重新分类之前必须完成以下步骤:
FDA 在《联邦公报》(Federal Register)上公布拟议命令(Proposed order),其中包括拟议的重新分类以及支持重新分类的有效科学证据摘要 FDA 召开医疗器械分类专家小组会议(Convene a device classification panel meeting) FDA 审议在公众意见征询期内收到的、针对该拟议命令提交至公开档案(Public docket)的意见(Comments)。
如果 FDA 提议将医疗器械从 II 类重新分类为 III 类,则现有科学证据(Scientific evidence)必须表明,仅依靠一般管制要求和特殊管制要求(General controls and special controls)不足以合理保证该医疗器械的安全性和有效性。反之,如果 FDA 提议将医疗器械从 III 类重新分类为 II 类,则现有科学证据(Scientific evidence)必须表明,一般管制要求和特殊管制要求(General controls and special controls)足以合理保证该医疗器械的安全性和有效性。最后,如果 FDA 提议将医疗器械从 III 类或 II 类重新分类为 I 类,则现有科学证据(Scientific evidence)必须表明,一般管制要求(General controls)足以合理保证该医疗器械的安全性和有效性。
根据第 513(f)(3) 条(Section 513(f)(3))对目前根据第 513(f)(1) 条(Section 513(f)(1))自动分类为 III 类的医疗器械进行重新分类(Reclassification)
根据第《联邦食品、药品和化妆品法案》 513(f)(3) 条(Section 513(f)(3) of the FD&C Act),对于修正案后根据第 513(f)(1) 条(Section 513(f)(1))自动归类为 III 类的医疗器械,美国食品药品监督管理局 FDA 可主动启动器械类型的重新分类程序,或响应相关方(如制造商或进口商)提出的将器械类型重新归类为 I 类或 II 类的重新分类申请(Petition)。要更改医疗器械类型的分类,该器械必须符合该类别医疗器械的定义要求。与上文所述的 513(e) 重新分类流程一样,要更改医疗器械的分类,拟议的新类别必须具备充分的监管控制措施(Sufficient regulatory controls),以合理保证该医疗器械在其预期用途内的安全性和有效性。如果 FDA 收到重新分类申请(Petition),FDA 将审查该申请是否存在任何妨碍其做出决定的缺陷。如果 FDA 认定该申请不存在此类缺陷,则 FDA 可基于正当理由(Good cause)将该申请转交给相应的医疗器械分类专业组(Device classification panel),由其审查相关信息并提出建议。FDA 在考虑相关信息后,将批准或拒绝该申请。如果 FDA 批准该申请,将发布一份命令,说明批准该申请(重新分类)的理由,并列明该命令所适用的医疗器械可能存在的健康风险(如有)。
监管控制(Regulatory Controls)
概述
美国《联邦食品、药品和化妆品法案》第 513 节(Federal Food, Drug, and Cosmetic Act, section 513)建立了基于风险的医疗器械分类系统。每种医疗器械根据其安全性和有效性所需的管制级别,被划分为三个监管类别之一:I 类(Class I)、II 类(Class II)或 III 类(Class III)。
随着医疗器械风险等级的提升,从 I 类到 III 类,为确保其安全性和有效性而所需的监管控制要求也随之加强。其中 I 类医疗器械受到的监管控制最少,而 III 类医疗器械受到的监管控制(Regulatory control)最为严格。
监管控制(Regulatory controls)规定了为合理保证医疗器械的安全性和有效性所必需的适当监管水平(Appropriate level of regulatory )。监管控制通过提供一致性要求,在可预见的监管负担或监督水平下,促进医疗器械的安全性和有效性。
每种医疗器械类别的监管控制要求包括:
I 类(中低风险):一般管制(General controls) II 类(中高风险):一般管制和特殊管制(General controls and Special Controls) III 类(高风险):一般管制和上市前批准 PMA(General controls and Premarket Approval (PMA))
总而言之,医疗器械是根据风险程度进行分类,而医疗器械的风险程度决定了监管控制的程度(Extent of regulatory controls)。
一般管制要求(General controls)
一般管制要求是《联邦食品、药品和化妆品法案》(FD&C Act)第 Sections 501, 502, 510, 516, 518, 519 条授权的监管要求,适用于所有类别医疗器械(即 I 类、II 类和 III 类),除非法规豁免。如果某医疗器械被豁免于某项一般管制要求,则该器械的分类法规中会说明该豁免情况。
例如,标签(Labeling)标识是一项基本的一般管制要求,21 CFR Part 801 详细规定了标签要求,其核心在于要求提供医疗器械的相关信息,以确保其安全有效使用。另一个例子是机构注册(Establishment registration),根据 21 CFR 807 的规定,机构必须每年向 FDA 进行注册。
FD&C Act 法案的以下章节对一般管制要求进行了说明:
501:掺假器械(Adulterated devices)
502:误标器械(Misbranded devices)
510:器械制造商注册(Registration of producers of devices)
机构注册和器械列名(Establishment registration and device listing) 上市前通知 510(K)(Premarket Notification (510k)) 再处理的一次性器械(Reprocessed single-use devices)
516:禁用器械(Banned devices)
518:通知和其他补救措施(Notifications and other remedies)
通知(Notification) 维修(Repair) 更换(Replacement) 退款(Refund) 补偿(Reimbursement) 强制性召回(Mandatory recall)
519:器械记录和报告(Records and reports on devices)
不良事件报告(Adverse event report) 器械追踪(Device tracking) 唯一器械识别系统(Unique device identification system) 移除和更正报告(Reports of removals and corrections)
520:关于管制供人类使用的医疗器械的一般规定(General provisions respecting control of devices intended for human use)
定制器械(Custom device) 受限器械(Restricted device) 良好生产规范要求(Good manufacturing practice requirements) 用于研究用途的器械的豁免(Exemptions for devices for investigational use) 被视为新药的器械的过渡性规定(Transitional provisions for devices considered as new drugs) 人道主义医疗器械豁免(Humanitarian device exemption)
特殊管制要求(Special controls)
特殊管制要求是针对 II 类医疗器械(Class II devices)的监管要求(Regulatory requirements)。因此 II 类医疗器械需同时遵守一般管制(General)和特殊管制(Special)的要求。FDA 将那些仅靠一般管制不足以合理保证其安全性和有效性,且有足够信息来建立特殊管制要求以提供此类保证的器械归类为 II 类医疗器械(Class II devices)。特殊管制要求(Special controls)通常是针对完善的医疗器械类型(Well-established device types)而制定的。由于 FDA 对这些医疗器械类型有充分的了解,能够确定一些确保其安全有效性的统一要求。
1976 年医疗器械修正案(Medical Device Amendments of 1976)(Pub. L. 94-295)中对 II 类(Class II)医疗器械的最初定义(Original definition)中,明确将性能标准(Performance standards)而非特殊管制(Special controls)作为 FDA 建立安全性和有效性合理保证的机制。1990 年安全医疗器械法案(The Safe Medical Devices Act of 1990)(Pub. L. 101-629)增加了“特殊管制”(Special controls),其中包括颁布性能标准(Promulgation of performance standards)、上市后监测(Postmarket surveillance)、患者登记(Patient registries)、制定并发布指南(含上市前通知中提交临床数据(Clinical data)的指南)以及 FDA 认为提供此类保证所必需的其他保障措施(Section 513(a)(1)(B) of the FD&C Act (21 U.S.C. § 360c(a)(1)(B))。
特殊管制要求(Special controls)的法规要求详见 CFR 的第 862 至 1050 部分(Code of Federal Regulations Parts 862 through 1050)。若某医疗器械类型存在特殊监管控制要求,则可在该器械特定法规的 Subsection B 子章节中查阅。

例如,21 CFR 876.5860 规定了高渗透性血液透析系统的监管要求。查阅该部分的子章节(Sub-section B of this section)可知,该医疗器械类型属于 Class II 类,并受该部分列出的 5 项特殊管制要求的约束。
特殊管制要求通常针对特定器械(Device-specific),包括:
性能标准(Performance standards) 上市后监测(Postmarket surveillance) 患者登记(Patient registries) 特殊标签要求(Special labeling requirements) 上市前数据要求(Premarket data requirements) 指南文件(Guidelines)
上市前批准 PMA(Premarket Approval)
根据美国联邦法律,III 类医疗器械(Class III)需要获得上市前批准 PMA(Premarket Approval)申请的批准。
在 1976 年《医疗器械修正案》(Medical Device Amendments of 1976)颁布之前未上市的医疗器械(称为修正案前器械)(Preamendments devices),将自动归类为 III 类医疗器械。此外,FDA 还将用于支持或维持人类生命或防止损害人类健康的器械,或可能存在潜在的不合理患病或受伤风险的器械划归为 III 类医疗器械,对于这些医疗器械,一般管制要求和特殊管制要求不足以合理保证器械的安全性和有效性,或者没有足够信息来做出此类判断。
医疗器械的一般管制要求
概述
一般管制要求(General Controls)是 1976 年 5 月 28 日通过的《联邦食品、药品和化妆品法案》《医疗器械修正案》(Medical Device Amendments)的基本条款(授权),为 FDA 提供了监管医疗器械的法律授权,以确保其安全性和有效性。修正案中的一般管制要求适用于所有医疗器械。涵盖与掺假(Adulteration)、误标(Misbranding)、医疗器械注册和列名(Device registration and listing)、上市前通知(Premarket notification)、禁用器械(Banned devices)、通知(包括修理、更换或退款)(Repair, replacement, or refund)、记录和报告(Records and reports)、受限器械(Restricted devices)以及良好生产规范(Good manufacturing practices)相关的规定。
一般管制要求条款的适用
除非另有豁免(Exempted),MDA 修正案的一般管制要求适用于所有医疗器械,无论其分类状态如何。
1,一般管制要求(General Controls)是《医疗器械修正案》(Medical Device Amendments)的基本授权,为 FDA 提供了监管医疗器械的手段,以确保其安全性和有效性。
2,一般管制要求(General Controls)适用于所有三个类别的医疗器械,但它是唯一适用于 I 类器械的管制措施。
3,I 类医疗器械(Class I)不适用于:
用于支持或维持生命; 对预防人类生命损害具有重要意义;或可能 造成潜在的不合理疾病或伤害风险
4,一般管制要求(General Controls)包括该法案中与以下方面有关的规定(Provisions):
掺假(Adulteration) 误标(Misbranding) 医疗器械注册和列名(Device registration and listing) 上市前通知(Premarket notification) 禁用器械(Banned devices) 通知及维修,更换和退款(Notification and repair, replacement, and refund) 记录和报告(Records and reports) 受限医疗器械(Restricted devices);以及 良好生产规范(Good Manufacturing Practices)
掺假(Adulteration)
医疗器械受 FD&C Act 法案第 Section 501 节关于掺假规定的约束。Section 501 的前 2 条规定(Provisions)在大多数情况下定义了掺假。如果医疗器械含有任何污秽、腐烂或分解的物质,或者其制备、包装或存放条件不卫生,则该医疗器械被视为掺假。FD&C Act 法案进一步规定,如果出现以下情况,则该医疗器械被视为掺假:
其容器全部或部分由任何有毒或有害物质构成; 其仅用于着色,含有不安全的色素添加剂;以及 其强度(Strength)与其声称的不符,或其纯度或质量低于其声称的纯度或质量。
在 FD&C Act 法案中新增《医疗器械修正案》时,在 Section 501 中新增了一些专用于医疗器械的合规法律。这些规定(Provisions)与修正案的其他部分直接相关,赋予 FDA 以下权力:控制性能标准;遵守上市前批准申请和产品开发方案要求;禁用器械;良好生产规范;以及研究用医疗器械豁免。这些条款(Sections)规定,如果出现以下情况,医疗器械也将被视为掺假:
受性能标准(Performance standard)约束,但不符合该标准的所有要求; 属于 III 类医疗器械(Class III),但不符合已获批准的上市前批准申请或产品开发方案完成通知的要求; 属于禁用医疗器械; 违反了良好生产规范的要求;或 不符合研究用医疗器械豁免 IDE(Investigational Device Exemption)的规定。
误标(Misbranding)
FD&C Act 法案第 Section 502 节中的关于误标的规定(Misbranding provisions)涵盖了药品和医疗器械标签要求的各个方面。其中许多条款(Provisions)同时适用于药品和医疗器械;然而,也有一些特定的误标规定(Misbranding provisions)仅适用于药品或医疗器械。以下列出了同时适用于药品和医疗器械的误标规定(Misbranding provisions):
药品和医疗器械误标规定(Drugs and Device Misbranding Provisions)
如果出现以下情况,药品或医疗器械将被视为误标:
1,标签是虚假和误导性的
2,包装上没有贴有包含以下内容的标签:
制造商、包装商或经销商的营业地点名称,以及 内容物的重量、尺寸或数量准确表述。
可以允许对小型包装进行合理的改动和豁免(Exemptions)。
3,任何文字、声明或其他必要信息未在标签上突出显示或未清晰表述,以便普通个人在通常的购买和使用条件下能够阅读和理解。
4,供人类使用并含有任何数量的麻醉或成瘾物质,除非其标签上标有该物质或衍生物的名称、数量或比例,以及 “警告--可能成瘾 ”(Warning - may be habit forming)的说明。
5,其标签未提供充分的使用说明。标签必须包含警告(Warnings),禁止在某些病理情况下使用,或禁止儿童使用,因为使用可能危害健康,或禁止使用不安全的剂量、方法或使用时间。在有必要保护使用者健康的情况下,必须提供充分的使用说明和警告。此规定可获得豁免(Exemptions)。“充分的使用说明”(Adequate directions for use)适用于非处方药品和医疗器械(over-the-counter drugs and device)。
6,如果按照标签上规定、推荐或建议的剂量、方式、频率或持续时间使用,则会对健康造成危害。
7,它不符合 FD&C Act 法案第 Section 706 条所列的色素添加剂规定(Provisions)。
修正案新增的医疗器械错误标签规定(Device Misbranding Provisions Added by the Amendments)
MDA 修正案新增了有关医疗器械误标的新规定。这些新条款(Provisions)规定,如果出现以下情况,则医疗器械将被认定为误标(Misbranded):
1,该医疗器械的既定名称(如有)、其在官方汇编中的名称、或任何常用名称,未以至少相当于任何专有名称或名称字体一半大小的醒目字体印刷。此规定可获豁免。
2,在任何州销售的受限医疗器械使用虚假或误导性广告,或违反 FD&C Act 法案第 Section 820(e) 条规定的受限医疗器械规定进行销售、分销或使用。
3,受限医疗器械制造商、包装商或分销商未能在所有广告或其他说明材料中说明以下内容:
真实陈述医疗器械的既定名称,并以醒目的方式打印,并且, 简要说明医疗器械的预期用途以及相关的警告,注意事项,副作用及矛盾之处。
4,该器械在未经 FDA 许可,未按提交 Section 510(k) 规定提交申请便已进行商业分销。
5,该器械受性能标准的约束,但未贴有该标准规定的标签。
6,未能或拒绝遵守 Section 518 条(通知和其他补救措施)(Notification and Other Remedies)规定的任何要求;未能提供 Section 518 条要求的任何材料或信息;或未能提供 Section 519 条(器械记录和报告)(Records and Reports on Devices)要求的任何材料或信息。
虚假或误导性标签(False or Misleading Labeling)
FD&C Act 法案规定,如果药品或医疗器械的标签(Labeling)“在任何方面存在虚假或误导性”,则该药品或器械被认定为误标(Misbranded)。“标签”(Labeling)包括标签和任何其他书面、印刷或图形材料,这些材料随附于医疗器械及其任何包装物或容器。操作和维修说明也被视为标签的一部分。标签必须包含足够的使用说明以及确保医疗器械安全有效使用所需的任何警告。
机构注册要求(Establishment Registration Requirements)
《医疗器械修正案》Section 510 条规定,医疗器械制造商和其他特定加工商必须向 FDA 注册其机构(Establishments),并向 FDA 列名在其机构内生产的所有医疗器械。再包装商(Repackers)、再贴牌商(Relabelers)和进口商也必须向FDA注册。
医疗器械列名要求(Device Listing Requirements)
Section 510 还规定,医疗器械制造商必须向 FDA 列名其生产或加工的所有医疗器械。该列名信息由 FDA 维护。
上市前通知要求(Premarket Notification Requirements)
FD&C Act 法案 Section 510(k) 条规定,有意销售医疗器械的制造商必须在将医疗器械上市前至少 90 天向 FDA 提交上市前通知(Premarket notification)510(k)。经 FDA 认定实质不等同 NSE 的医疗器械(Non equivalent device)必须拥有已获批准的上市前批准申请 PMA(Premarket approval),或重新归类为 I 类或 II 类后才能上市。
禁用医疗器械(Banned Devices)
FD&C Act 法案 Section 516 条授权 FDA 禁止那些存在重大欺骗性或不合理且重大致病或致伤风险的器械。禁用某种医疗器械的程序(Procedures)如下所述。如果 FDA 根据所有现有的数据和信息,并在咨询相关分类专业组(Classification panel)后,认定某种拟供人类使用的医疗器械存在上述欺骗性或致病或致伤风险,且无法通过更改标签予以纠正,则 FDA 可在《联邦公报》(Federal Register)上发布一项拟议法规(Proposed regulation),禁止该医疗器械(Ban the device)。如果可以通过更改标签来纠正欺骗或风险,则 FDA 必须将欺骗或风险、纠正欺骗或风险所需的标签更改以及作出更改的期限通知责任人(Responsible person)。如果标签(Labeling)未在规定的时间内以规定的方式更改,则该 FDA 可会发布拟议法规。在为所有相关方提供就该提案(Proposal)进行非正式听证的机会后,FDA 将确认、修改或撤销拟议的法规。如果提案获得确认或修改,FDA 将发布最终法规(Final regulatio),禁止该医疗器械。在这种情况下,该器械自最终法规发布之日起及之后将不能再合法上市销售,除非获得已获批准的研究用医疗器械豁免(Investigational device exemption)。如果拟议法规被撤销(Revoked),FDA 将在《联邦公报》上发布相关通知。
通知和其他补救措施(Notification and Other Remedies)
FD&C Act 法案第 Section 518 条涉及通知和其他补救措施(Notification and Other Remedies),以保护公众免受故障或欺诈性器械的侵害。
Section 518 条款的目的
Section 518 的主要目的是保护公众健康。Section 518 为 FDA 提供了一种途径,确保消费者和其他用户手中的危险产品得到维修、更换或退款(Repaired, replaced, or refunded)。除了保护公众健康这一目的外,Section 518 还为消费者在购买存在不合理风险的缺陷医疗器械时提供了经济赔偿的程序。
通知 518(a)(Notification 518(a))
根据 FD&C Act 法案的这一条款,FDA 可以要求制造商或其他相关人员通知所有开具或使用该器械的医疗专业人员以及任何其他人员(包括制造商、进口商、分销商、零售商和器械用户)使用违规器械所导致的健康风险,以便减少或消除这些风险。
门槛性要求(Threshold Requirements)
如果出现以下情况,FDA 可以下令通知:
某医疗器械存在对公众健康造成重大损害的不合理风险; 有必要发出通知以消除风险;以及 根据 FD&C Act 法案,没有更切实可行的方法来消除风险。
程序(Procedures)
通知令(Notification order)的程序很简单,只需要事先与需要提供通知的人员进行协商即可。
维修,更换或退款规定518(b) (Repair, Replacement, or Refund Provisions 518(b))
Section 518(b) 授权 FDA 在提供非正式听证机会(Informal hearing)后,命令制造商、进口商或分销商修理、更换或退还存在不合理健康风险的医疗器械的购买价格。
基本准则(Basic Criteria)
如果 FDA 在非正式听证会后确定以下情况,则可以下令修理、更换或退款(3-R):
该器械存在对公众健康造成重大损害的不合理风险; 该器械的设计和制造不符合当时的技术水平(State of the art); 风险并非由制造商、进口商、分销商或零售商以外的人员在安装、维护、维修或使用该器械时疏忽造成;并且 仅通知是不够的,必须进行维修、更换或退款。
程序(Procedures)
维修,更换或退款(The procedures for repair, replacement, or refund)程序复杂,如果 FDA 和制造商或其他责任人无法就风险应对方案达成一致,可能会导致多次下达命令、召开监管听证会并造成严重延误。FDA 必须考虑其他可行的替代方案。通知令(Notification orders)以及维修、更换或退款令(Repair, replacement, or refund orders)均由 FDA 酌情决定。在下达通知令之前,FDA 必须确定根据 FD&C Act 法案,没有更切实可行的方法来消除风险。虽然 FDA 没有要求在下达维修、更换或退款令之前必须做出这样的决定,但 FDA 必须确定,在下达维修、更换或退款令之前,仅仅通知是不够的。
FDA 对 Section 518 的替代方案如下:
法律行动(扣押、禁令、起诉); 法规(例如,禁止或限制销售、分销或使用);以及 召回(根据 FDA 的召回规定)。
器械记录和报告(Records and Reports on Devices)
FD&C Act 法案 Section 519 节授权 FDA 颁布法规,要求器械制造商、进口商和分销商保存记录和报告,以确保器械没有掺假(Adulterated)或误标(Misbranded)。
根据 Section 519 颁布的记录和报告法规:
不得对制造商、进口商或分销商施加过度负担(Unduly burdensome)的要求; 必须说明要求提供报告或信息的程序的理由和目的; 必须说明提交报告或信息的理由和目的; 不得要求披露任何患者的身份; 不得要求制造商、进口商或分销商保存或提交其未持有的报告或信息。
记录和报告要求(Records and reports requirements)不适用于:
仅在其专业实践过程中开具或管理器械的执业人员; 仅用于研究或教学的器械制造商或进口商;以及 其他受法规豁免的人员。
受限器械(Restricted Devices)
根据《联邦食品、药品和化妆品法案》第520(e)条(Section 520(e)),如果无法合理保证某种医疗器械的安全性和有效性,FDA 有权限制其销售、分销或使用(Sale, distribution, or use)。受限器械只能在获得执业医师口头或书面授权后,或在法规规定的条件下销售。例如,心脏起搏器(Cardiac pacemakers)和心脏瓣膜(Heart valves)等医疗器械需要获得执业医师(Practitioner)的授权。
质量体系规范,良好生产规范(Quality System Regulation, Good Manufacturing Practices)
MDA 修正案第 520(f) 条(Section 520(f) of the Amendments)授权 FDA 颁布法规,要求医疗器械制造、包装、储存和安装所使用的方法以及所使用的设施和控制符合现行良好生产规范 (GMP)。
FDA 医疗器械常见问题 FAQ
我如何知道我的产品是否受 CDRH 监管?
FDA 负责监管医疗器械,以确保其安全性和有效性。FDA 下属的医疗器械与放射健康中心 CDRH(Center for Devices and Radiological Health)是负责执行此项监管的部门。FDA 监管医疗器械的法律依据是《联邦食品,药品与化妆品法案》(FD&C Act)(Federal Food Drug & Cosmetic Act)。
要使医疗器械获得批准,需要遵循哪些程序(Protocol)?
从 CDRH 获得上市许可(Marketing Clearance)需三个步骤。具体如下:
医疗器械上市流程(Marketing process)的第一步(STEP ONE)确保你希望销售(Wish to market)的产品是医疗器械,也就是说它是否符合《联邦食品、药品和化妆品法案》(FD&C Act)Section 201(h) 中对医疗器械的定义。例如,该产品可能是受 FDA 其他部门(而非医疗器械与放射健康中心 CDRH)(Center for Devices and Radiological Health)监管的药品或生物制品(Drug or biological product),并且 FD&C Act 法案对此类产品有不同的规定。或者你的产品虽属医疗器械,但同时也是电子辐射发射产品(Electronic radiation emitting product),需满足额外要求(Additional requirements)。
第二步(STEP TWO)是确定 FDA 将如何对你的医疗器械进行分类,即该医疗器械可能属于三个类别中的哪一类。除非获得豁免(Unless exempt),否则 FDA 将对你的医疗器械进行分类(Classify your device)。分类(Classification)确定了确保医疗器械安全性和有效性所需的监管控制级别(The level of regulatory control)。最重要的是,除非获得豁免,医疗器械的分类将确定制造商(Manufacturer)必须完成的上市流程(Marketing process),上市前通知(Premarket notification)510(k) 或上市前批准 PMA(Premarket approval),以获得FDA的上市许可/批准(FDA clearance/approval for marketing)。
第三步(STEP THREE)收集提交上市申请(Submit a marketing application)并获得 FDA 上市许可(FDA clearance to market)所需的数据和/或信息。对于某些 510(k) 申报和大多数 PMA 申请,需要提供临床性能数据(Clinical performance data)才能获得上市许可(Clearance to market)。在这些情况下,除了获得上市许可(Marketing clearance)外,试验(Trial)的进行还必须符合 FDA 的研究用医疗器械豁免 IDE(Investigational Device Exemption)规定。
准备和提交上市前通知 510(k) 申报的步骤是什么?
医疗器械的上市前通知 510(k) 申报由美国食品药品监督管理局下属的医疗器械与放射健康中心 CDRH(Center for Devices and Radiological Health)负责审查和处理(Reviewed and processed)。CDRH 下属的医疗器械评估办公室 ODE(Office of Device Evaluation)和体外诊断器械评估与安全办公室 OIVD(Office of In Vitro Diagnostic Device Evaluation and Safety)负责处理和审查 510(k) 申报,以获得美国市场的上市许可(Marketing clearance in the U.S.)。
这些办公室的分支机构按医学科学学科进行组织。ODE 和 OIVD 的评审人员包括生物医学工程师、医生、微生物学家、化学家等,他们负责对 510(k) 申报和其他研究类(研究用医疗器械豁免 IDE)(Investigational Device Exemption)以及上市类(Marketing)(上市前批准)(Premarket Approval)申请进行科学审查(Scientific reviews)。这些评审人员通常被称为“评审员”(Reviewers),他们的专业判断将决定新器械(New device)是否符合“实质等同”(SE)(Substantially equivalent)或“非实质等同”(NSE)(Not substantially equivalent)。
我如何了解 FDA 将如何对我的设备进行分类?
要确定医疗器械的分类以及是否存在任何豁免(Exemptions),需要查找适用于该医疗器械的分类法规编号。有两种方法:直接访问 FDA 的医疗器械产品分类数据库并搜索器械名称的部分内容;或者,如果你知道医疗器械所属的医疗器械分类专业组(Device panel)(Medical specialty)医学专业领域,则可直接访问该专业组(Panel)的列表(Listing),从而确定你的医疗器械产品及其对应的监管法规。
我的医疗需要贴标签吗?标签上应该包含哪些信息?
医疗器械的一般标签要求(General labeling requirements)载于 21 CFR Part 801 部分中。这些法规(Regulations)规定了所有医疗器械的最低要求。












