




【美国进口清关】FDA 医疗器械数据库

 2668
2668

一键完成百千笔付款,超低费率+极速到账,一年轻松帮你省下数十万。
1938 年《联邦食品、药品和化妆品法案》(FD&C Act)是授权 FDA 监管医疗产品的主要授权立法,医疗器械必须满足 FDA 的监管要求才能在美国上市销售。
FDA 医疗器械监管概述
FDA 是美国联邦政府中历史最悠久的综合性消费者保护机构(Comprehensive consumer protection agency)。
1938 年《联邦食品、药品和化妆品法案》(FD&C)(Federal Food, Drug, and Cosmetic Act of 1938)的通过取代了 1906 年的《纯净食品和药品法案》(Pure Food and Drug Act of 1906),医疗器械(Medical devices)首次受到全面监管。
20 世纪 70 年代,美国国会通过了《1976 年医疗器械修正案》(MDA)(Medical Device Amendments of 1976),以回应公众对医疗器械加强监督的渴望。MDA 为医疗器械的分类建立了一个基于风险的框架,为医疗器械进入市场以及医疗器械临床试验建立了监管途径,并制定了多项上市后要求,包括制造商注册和 FDA 医疗器械列名、良好生产规范 GMP 以及不良事件报告。
美国食品药品监督管理局 FDA 采用基于风险的分级管理方法(Risk-based, tiered approach)来对医疗器械进行监管,FDA 根据风险等级和为合理保证器械的安全性和有效性所需的监管控制程度(Level of regulatory controls)对医疗器械进行分类。
医疗器械分为三类监管类别(Regulatory classes):I 类(Class I)、II 类(Class II)和 III 类(Class III)。I 类器械通常对患者和/或用户的风险最低,而 III 类器械的风险最高。风险最高的器械(III 类),例如机械心脏瓣膜和植入式输液泵,通常需在上市前(Before marketing)获得 FDA 对上市前批准 PMA 申请(Premarket approval application)的批准(Approval)。制造商(Manufacturers)若要获得 FDA 对这类器械的批准,必须通过充分有效的科学证据证明,该器械在预期用途内具有合理的安全性和有效性保障。
通常情况下,FDA 会在确认某中等风险医疗器械(Moderate-risk medical devices)(II 类)(Class II)(例如透析设备和多种类型导管)与无需上市前批准(Premarket approval)的已合法上市(Legally marketed)的对比器械(Predicate device)实质等同(Substantially equivalent)后,批准其上市(Clears)。II 类医疗器械(Class II)通常受到特殊管制要求的约束(Subject to special controls),包括针对该器械的特定测试或标签要求。
对用户造成伤害风险较低的器械(I 类)(Class I)(例如非电动吸乳器、弹性绷带、压舌板和检查手套)仅受一般管制要求的约束(Subject to general controls only),且大多数不受上市前通知要求(Premarket notification requirements)的约束。
FD&C Act 法案中的条款(Provisions),即监管要求(Regulatory requirements),规定了 FDA 对这些产品的监管程度。
为了履行 FD&C Act 法案中适用于医疗器械的规定,FDA 制定、发布并实施相关法规。这些法规(Regulations)最初在《联邦公布》FR 上发布,征询公众意见。随后,最终法规(Final regulations)每年都会被纳入或汇编于(Placed or codified into)《联邦法规汇编》CFR(Code of Federal Regulations),FDA 的大部分医疗器械(Medical device)和发射辐射产品(Radiation-emitting product)法规都载于《联邦法规汇编》第 21 篇第 800-1299 部分(Title 21 CFR Parts 800-1299)。这些编入《联邦法规汇编》的最终法规(Final regulations)涵盖了医疗器械的设计(Design)、临床评估(Clinical evaluation)、制造(Manufacturing)、包装(Packaging)、标签(Labeling)和上市后监测(Post market surveillance)等各个方面。
FDA 医疗器械数据库
医疗器械产品分类数据库
医疗器械产品分类数据库(Product Classification):https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm
FDA 对在美国销售的医疗器械进行监管,以确保其安全性和有效性。
医疗器械产品分类数据库列出了 6000 多种受 FDA 医疗器械和放射健康中心 CDRH(Center for Medical Devices and Radiological Health)监管的医疗器械,及其相关分类、产品代码、FDA 上市前审查机构及其他监管信息。
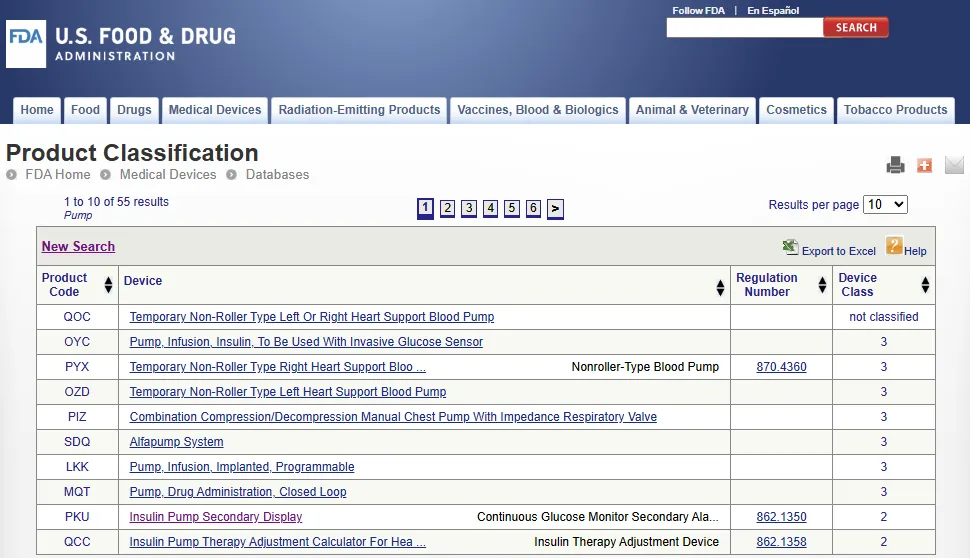
医疗名称和产品代码用于标识 FDA 规定的器械通用类别(Generic category)。
分配给器械的产品代码基于 21 CFR 第 862-892 条(21 CFR Parts 862-892)规定的医疗器械产品分类。
510(k) 上市前通知数据库
510(k) 上市前通知数据库(510(k) Premarket Notification):https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/pmn.cfm
510(k) 是向 FDA 提交的上市前申请(Premarketing submission),旨在证明拟上市的医疗器械与已合法上市且无需上市前批准 PMA(Premarket approval)的医疗器械(Section 513(i)(1)(A) FD&C Act)具有同等的安全性和有效性,即实质等同 SE(Substantially equivalent)。
医疗器械制造商(Medical device manufacturers)如计划首次将某医疗器械投入商业分销(Commercial distribution),或将某医疗器械重新推向市场且该器械将进行重大变更或修改(Significantly changed or modified),以致其安全性或有效性(Safety or effectiveness)可能受到影响,则必须提交上市前通知(Premarket notification)510(k)。
可向给 FDA 提交的上市前通知 510(k) 有三种类型:传统型(Traditional)、特殊型(Special)和简化型(Abbreviated)。除非该医疗器械符合 510(k) 豁免条件,否则必须在上市前至少 90 天向 FDA 提交 510(k)(上市前通知)(Premarket notification)。
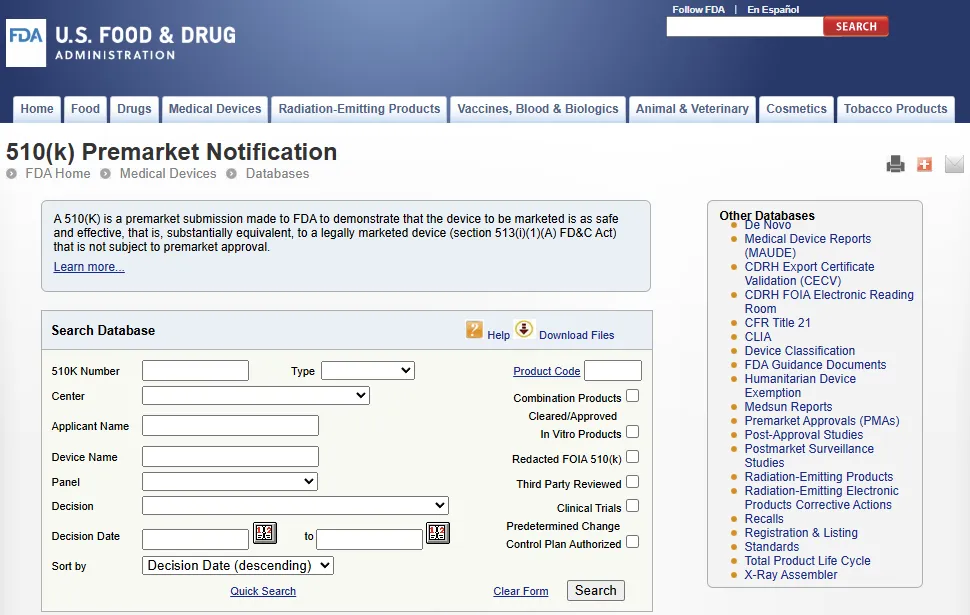
510(k) 上市前通知数据库每周更新一次,可按 510(k) 编号、申请方、器械名称或 FDA 产品代码进行搜索。
系统采用精确匹配检索, FDA 建议在按器械名称(Device Name)进行搜索时,在输入框中仅填写一个描述性关键词,
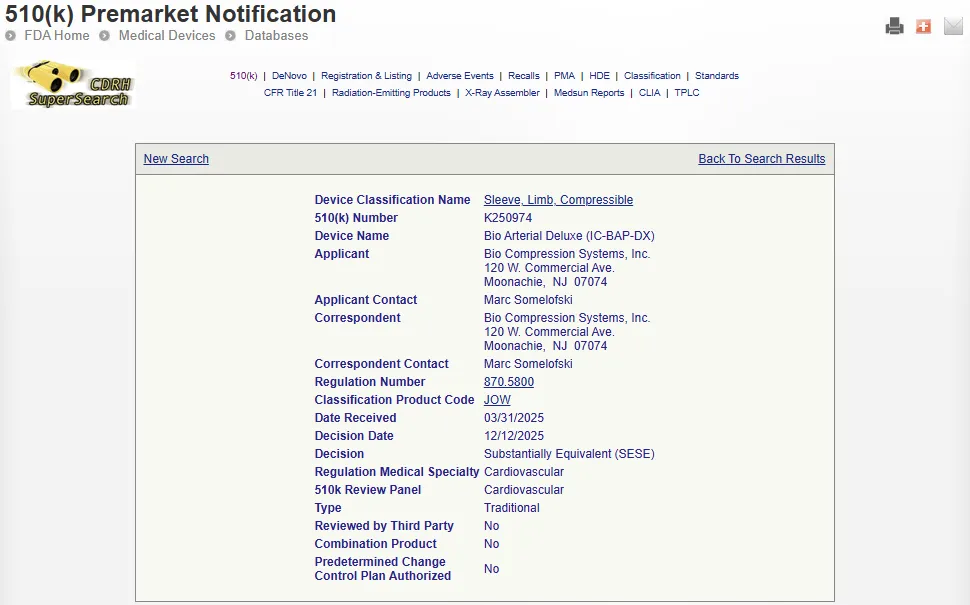
搜索查询将以以下格式从数据库中返回信息:
Device Classification Name:器械分类名称 510(k) Numbe:510(k) 编号 Device Name:医疗器械名称 Applicant:申请方 Applicant Contact:申请方联系人 Correspondent:通讯员 Correspondent Contact:通讯员联系人 Regulation Number:法规编号 Classification Product Code:分类产品代码 Date Received:收到日期 Decision Date:决定日期 Decision:决定 Regulation Medical Specialty:医疗器械法规医学专业分类 510k Review Panel:510(k) 评审专业组 Summary:摘要 Type:510(k) 类型 Reviewed by Third Party:是否第三方审核 Combination Product:是否组合产品 Predetermined Change Control Plan Authorized:预先确定的变更控制计划是否已获批准
另外,需注意的是,部分企业名称(Firm names)可能并非当前的 510(k) 申请持有者,当 510(k) 申请的所有权从原申请人转移至其他公司时,上市前通知数据库不会更新。
医疗器械机构注册&器械列名数据库
机构注册&器械列名数据库(Establishment Registration & Device Listing)https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfRL/rl.cfm
参与生产和分销拟在美国使用的医疗器械的机构的所有者或经营者,需要每年向 FDA 进行注册。通常,需要向 FDA 注册的机构还需列名其生产的器械以及在这些器械上进行的活动。如果某医疗器械在美国上市前需要提交上市前申请,则所有者/经营者还应提供该医疗器械的 FDA 上市前申请编号(Premarket submission number)(510(k)、De Novo、PMA、PDP、HDE)。
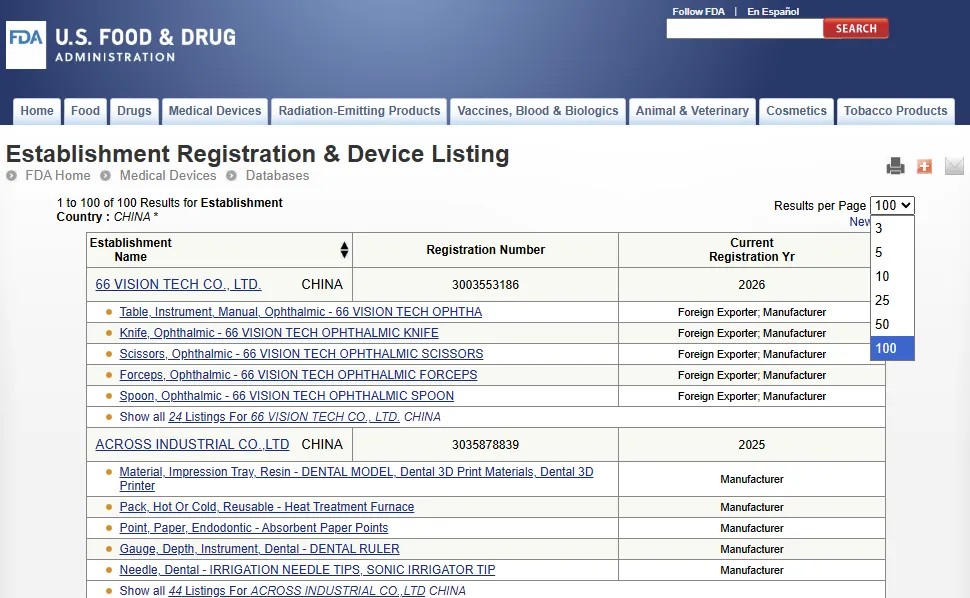
每次搜索最多只能返回 100 个不同的机构(Establishments)。
另外,需注意的是,医疗器械机构的注册、注册号的分配或医疗器械的列名并不以任何方式表示该机构或其产品获得 FDA 的批准。
医疗器械召回数据库
FDA 医疗器械召回数据库(Medical Device Recalls):https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfres/res.cfm
召回(Recall)是指企业(Firm)移除或纠正(Removal or correction)已上市产品(包括其标签和/或宣传材料)的行为,这些产品被美国食品药品监督管理局 FDA 认定违反其监管法规。如果产品未被召回,FDA 将采取法律行动,例如扣押或其他行政或民事诉讼措施。
召回可以由企业主动发起,也可以应 FDA 要求执行,或根据法定授权由 FDA 下令强制执行。召回不包括市场撤回(Market withdrawal)或库存回收(Stock recovery)。市场撤回指企业对已分销产品的撤回或整改(Removal or correction),涉及轻微违规(不构成 FDA 法律追责)或无违规情形。库存回收(Stock recovery)指对尚未投放市场或仍处于制造商直接控制下的医疗器械进行的纠正或移除(Correction or removal)措施,即该器械位于制造商所有或控制的场所内,且涉及纠正或移除行动的批次、型号、代码或其他相关单元的任何部分均未投放市场销售或投入使用。
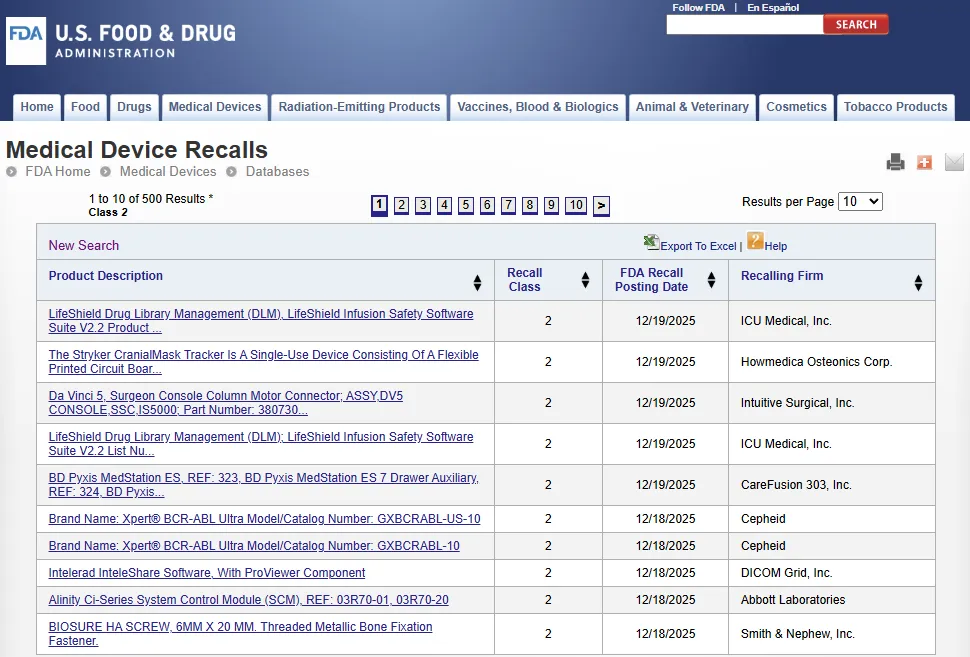
该数据库包含自 2002 年 11 月 1 日以来归类的医疗器械召回(Medical Device Recalls)信息。虽然随着新产品的上市,召回数据库会频繁更新,但它每月都会与 TPLC 数据合并。
自 2017 年 1 月起,该数据库可能还会包含企业(Firm)在 FDA 审查前启动的纠正或撤回措施(Correction or removal actions)。当 FDA 认定存在违规行为并将该行动归类为召回(Recall)时,状态将进行更新;召回终止时状态亦将再次进行更新。
FDA 的召回分类(FDA recall classification)可能发生在召回医疗器械产品的企业实施召回并向客户通报之后。因此,召回信息发布日期(“创建日期”)(Create date)指的是 FDA 对召回进行分类的日期,并不一定意味着该召回事件为新近发生。
Devices@FDA 数据库
Devices@FDA 数据库:https://www.accessdata.fda.gov/scripts/cdrh/devicesatfda/index.cfm
Devices@FDA 是一个收录了美国食品药品监督管理局 FDA 已许可和批准(Cleared and approved)医疗器械信息的目录(Catalog),该数据库每周更新,
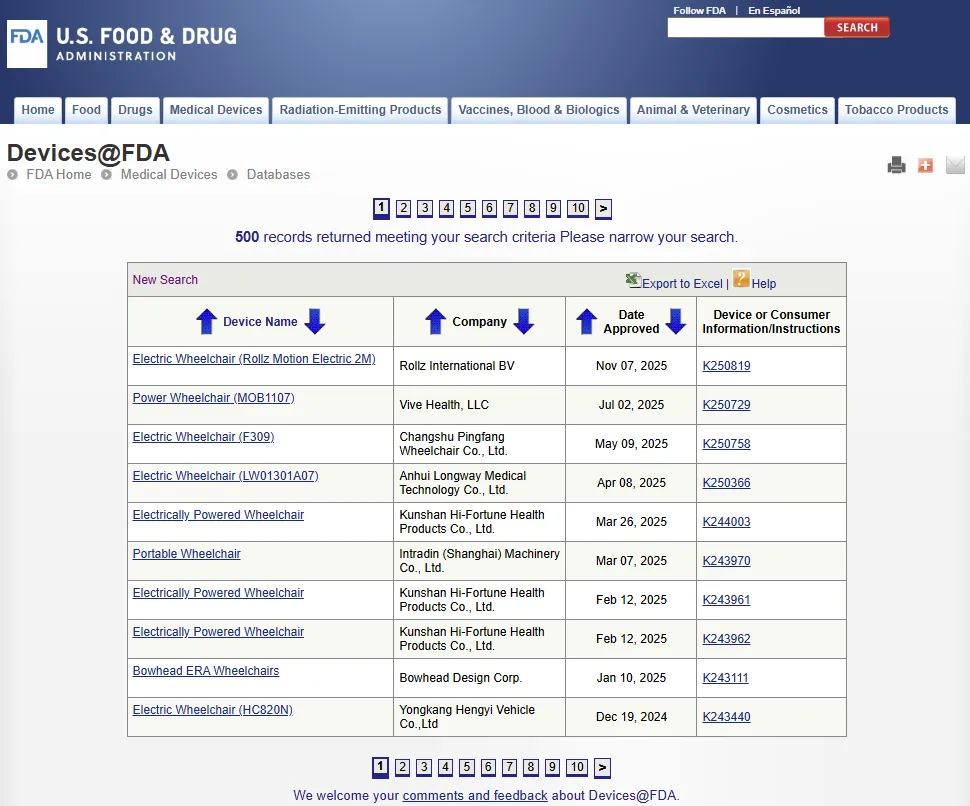
它包含医疗器械概要信息、制造商、批准日期(Approval date)、用户说明(User instructions)和其他消费者信息的链接。Devices@FDA 会搜索以下数据库:上市前通知 510(k) 和上市前批准 PMA。
发射辐射电子产品代码数据库
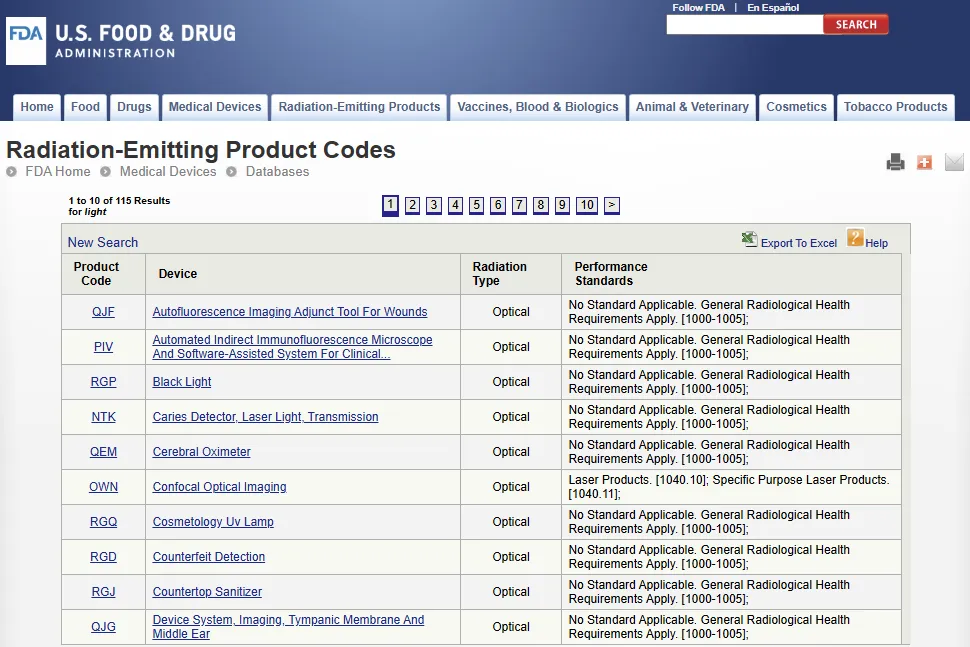
发射辐射电子产品代码数据库(Radiation Emitting Electronic Product Codes):https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD_rh/TextSearch.cfm
该数据库包含 Center 为所有发射辐射产品(Emit radiation)(包括医疗与非医疗产品)制定的产品名称及相关信息,
产品全生命周期 TPLC 数据库
产品全生命周期 TPLC(Total Product Life Cycle)数据库:https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfTPLC/tplc.cfm
TPLC 数据库整合了来自多个 CDRH 数据库(数据源)的数据,以呈现医疗器械上市前和上市后活动的综合记录。包括:
上市前批准 PMA(Premarket Approvals) 人道主义医疗器械豁免 HDE(Humanitarian Device Exemptions) 新分类申请(De Novo Classification Requests) 上市前通知 510(k)(Premarket Notifications) 不良事件(Adverse Events)(通过医疗器械制造商及使用机构不良事件数据库报告)(Manufacturer and User Facility Device Experience (MAUDE));以及 医疗器械召回(Medical Device Recalls)
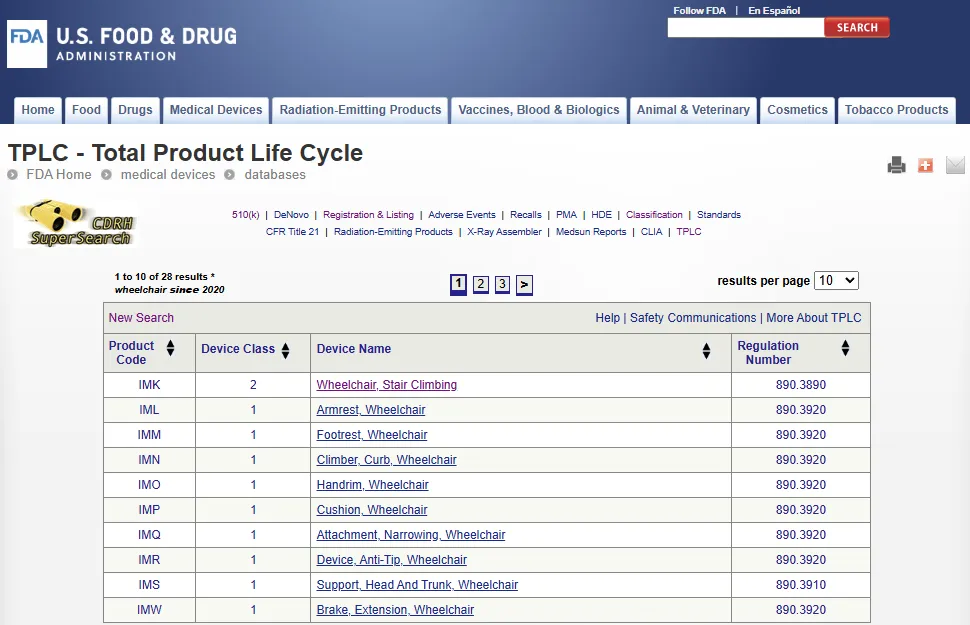
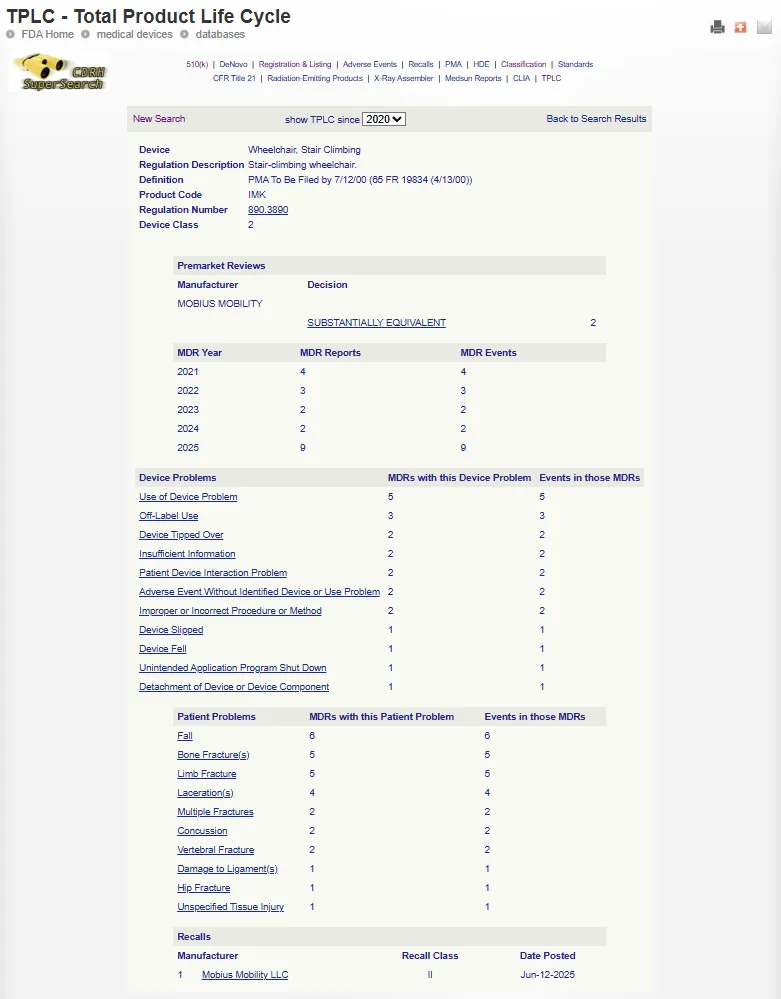
TPLC 数据库通过产品代码(Procode)整合数据,产品代码是一个与医疗器械通用类别(Generic type of medical device)相关的三字母代码(Three-letter code)。只有包含产品代码的数据集才会被纳入 TPLC 数据库。由于并非所有数据源中的条目都关联了产品代码,因此未关联代码的这些数据将被排除在 TPLC 数据库之外。
TPLC 报告包含数据库信息的统计数据(例如召回次数、上市前申报次数 Premarket submissions),有关统计事件的详细信息可通过页面上的链接查看。
医疗器械制造商及使用机构不良事件数据库
医疗器械制造商及使用机构不良事件数据库 MAUDE(Manufacturer and User Facility Device Experience Database):https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfMAUDE/search.CFM
《联邦食品、药品和化妆品法案》第 519 条(Section 519 of the FD&C Act)和医疗器械报告 MDR(Medical Device Reporting)法规 21 CFR Part 803 要求制造商若收到涉及医疗器械的故障、严重伤害或死亡事件的投诉,须向美国食品药品监督管理局 FDA 报告该事件。
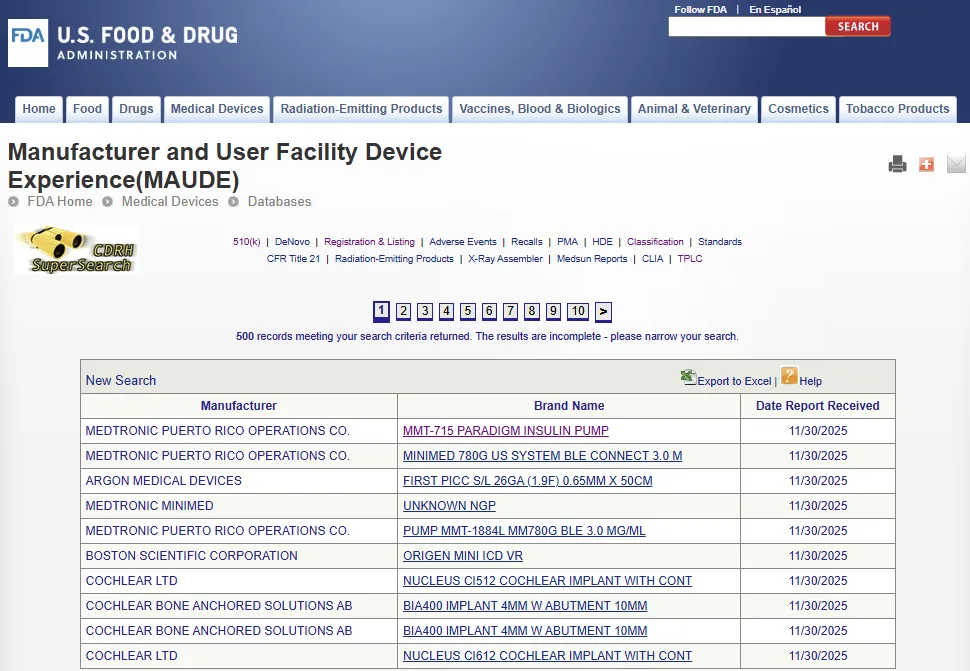
MAUDE 数据库(医疗器械不良事件报告数据库)收录了强制报告方(Mandatory reporters)(制造商、进口商和医疗器械使用机构)和自愿报告方(Voluntary reporters)(如医疗保健专业人员、患者和消费者)向 FDA 提交的医疗器械报告(Medical device reports),数据库每月更新一次。
该数据涵盖自 1993 年 6 月以来的所有自愿报告(Voluntary reports)、自 1991 年以来的用户机构报告(User facility reports)、自 1993 年以来的分销商报告(Distributor reports)以及自 1996 年 8 月以来的制造商报告(Manufacturer reports)。
高级搜索允许用户按特定字段(例如器械品牌名称、制造商等)在特定时间范围内进行搜索。
医疗器械公认共识标准数据库
医疗器械公认共识标准数据库(Recognized Consensus Standards):https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/search.cfm
该数据库提供最新的自愿性共识标准列表,FDA 将接受符合这些标准的医疗器械提交符合性声明。
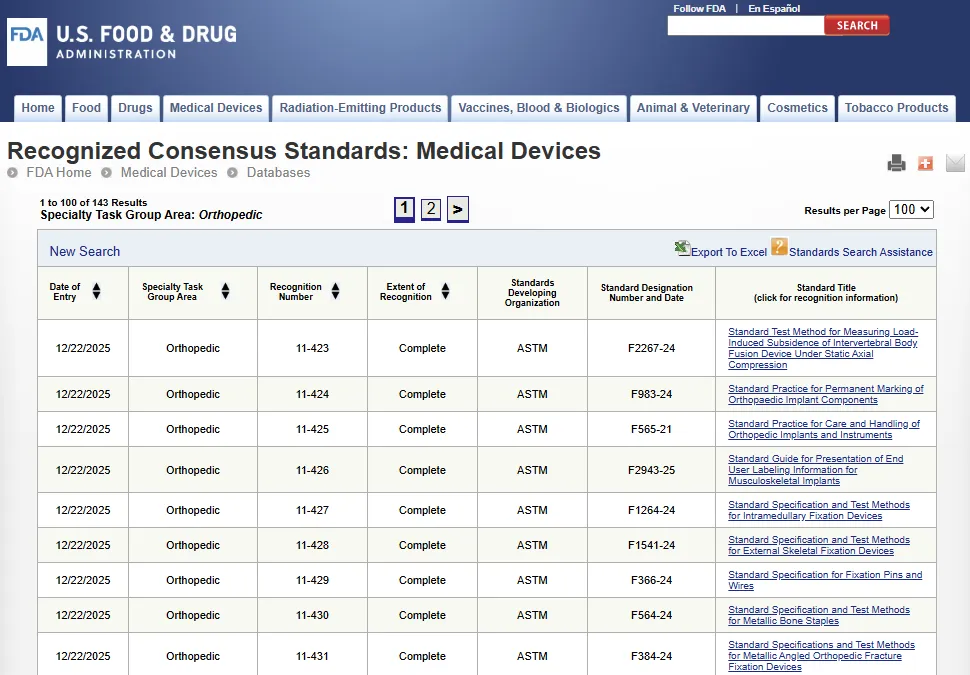
自愿性共识标准(Voluntary consensus standard)是由美国国内和国际标准制定组织 SDOs(Standards Development Organizations)根据严格的共识原则制定或采用的标准。FDA 决定认可某项标准(Standard)后,会在联邦公报(Federal Register)正式发布之前更新在线数据库,以反映该决定。
FDA 未认可标准数据库
FDA 未认可标准数据库(FDA's Non-Recognized Standards Database):https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/nr_results.cfm
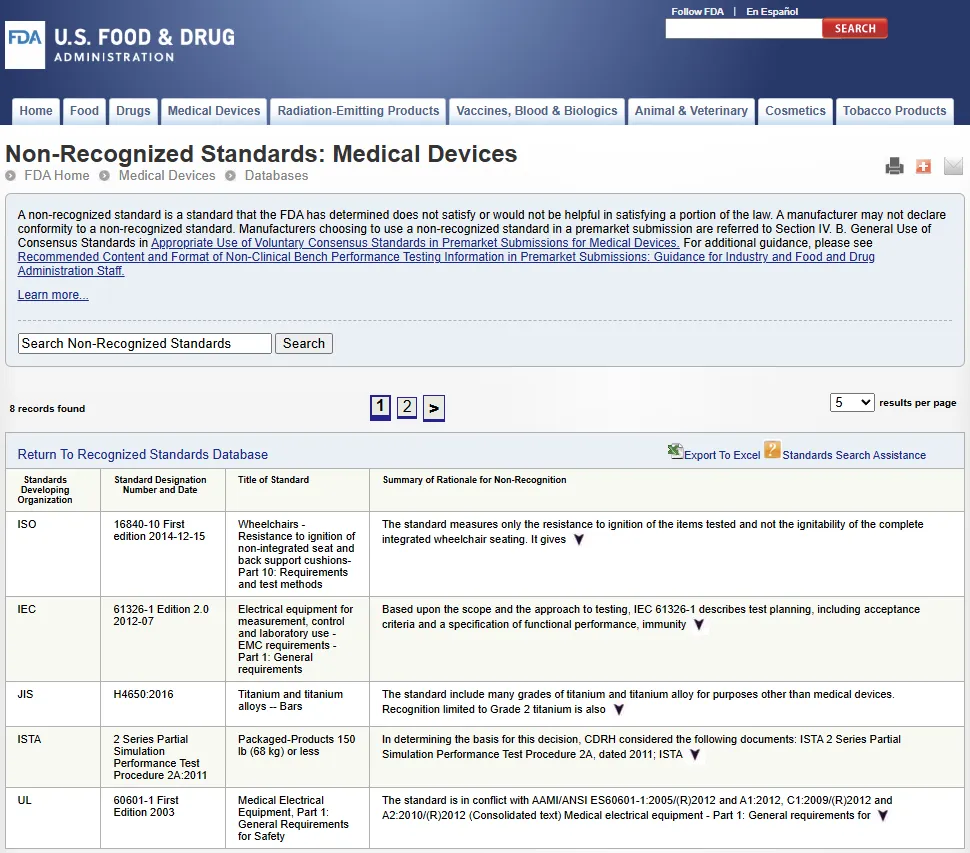
FDA 的《未认可标准数据库》中列出了所有被认定为未获认可的标准(List of FDA Non-Recognized Standards)及其未获认可的理由。
FDA 未认可标准(Non-Recognized Standard)是指 FDA 认定不符合或无法有效满足《联邦食品、药品和化妆品法案》(FD&C Act)(包括 1997 年《FDA 现代化法案》FDAMA)(FDA Modernization Act of 1997)和 2016 年《21 世纪治愈法案》(21st Century Cures Act of 2016)部分条款或相关法规的标准。制造商不得声明其产品符合这些未认可标准。
医疗器械唯一器械标识数据库 AccessGUDID
医疗器械唯一器械标识数据库 AccessGUDID:https://accessgudid.nlm.nih.gov/
FDA 的医疗器械唯一器械标识数据库 GUDID(Global Unique Device Identification Database)包含向 FDA 提交的有关具有唯一器械标识符 UDI(Unique Device Identifiers)的医疗器械的关键器械识别信息(Key device identification information)。例如标签上的器械标识符、器械名称、公司名称、MR 磁共振安全状态和上市前提交(Premarket submission)编号等。
上市前批准 PMA 数据库
上市前批准 PMA(Premarket Approvals)数据库:https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMA/pma.cfm
上市前批准 PMA(Premarket Approval)是 FDA 评估 III 类医疗器械的安全性和有效性的科学和监管审查流程。
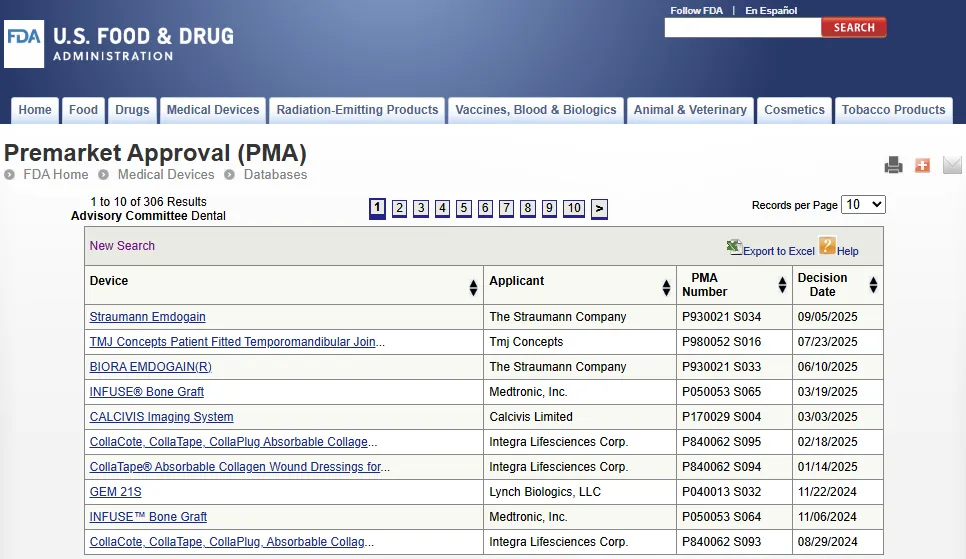
PMA 是 FDA 要求的最严格的医疗器械上市申请类型。获得批准(Approved)的 PMA 申请实际上是 FDA 授予申请方销售特定医疗器械的专属销售许可(Private license)。
该数据库每月更新一次,列出所有通过上市前批准 PMA 途径获准的新型或高风险医疗器械(New or high-risk medical devices)。
人道主义医疗器械豁免 HDE 数据库
人道主义医疗器械豁免 HDE 数据库(Humanitarian Device Exemption):https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfhde/hde.cfm
该数据库可查看已通过人道主义医疗器械豁免审查流程(HDE review process)获得批准的医疗器械。
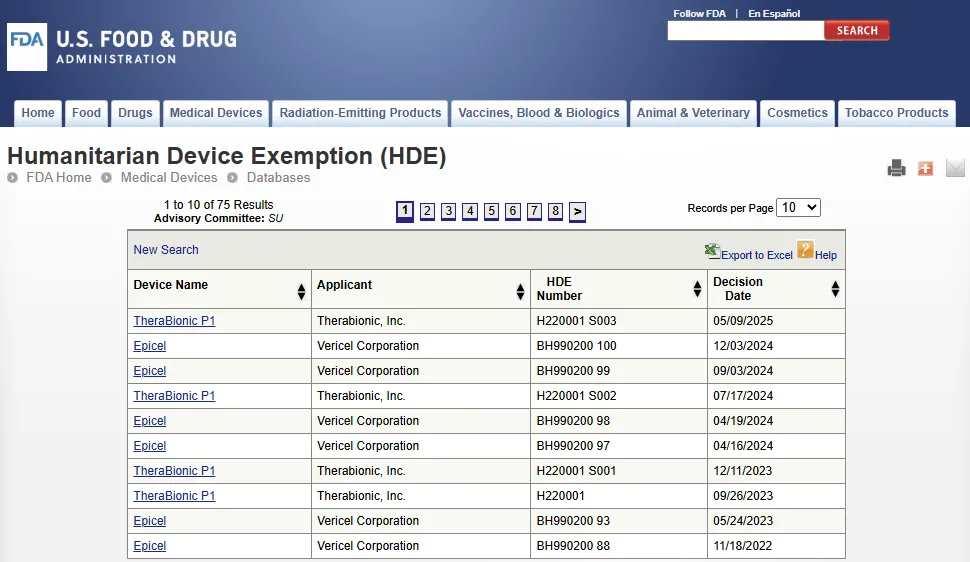
美国国会(Congress)在 1990 年《安全医疗器械法案》(Safe Medical Devices Act of 1990)中加入了一项条款(Provision),旨在为针对影响少数(罕见)人群的疾病或病症的产品建立监管途径(Regulatory pathway),此即人道主义医疗器械豁免 HDE 计划。
1996 年 6 月 26 日,美国食品药品监督管理局 FDA 发布了一项最终规则(Final rule),以落实 1990 年《安全医疗器械法案》(Safe Medical Devices Act of 1990)中关于人道主义用途医疗器械 HUDs(Humanitarian use devices)的规定(Provisions)。该法规(Regulation)于 1996 年 10 月 24 日生效。
人道主义用途医疗器械 HUD(Humanitarian Use Device)是指旨在通过治疗或诊断(Treating or diagnosing)每年在美国影响人数少于 8000 人的疾病或病症(Disease or condition)而使患者受益的医疗器械(Section 3052 of the 21st Century Cures Act)(Pub. L. No. 114-255)。针对小众患者群体的疾病或病症,医疗器械制造商的研发成本可能超过其市场收益。因此,FDA 制定并发布了这法规,旨在激励针对此类患者群体疾病的治疗或诊断医疗器械的研发。
该法规允许提交人道主义医疗器械豁免 HDE(Humanitarian device exemption)申请(Application),其形式和内容与上市前批准 PMA(Premarket approval)申请类似,但可豁免 PMA 的有效性要求(Effectiveness requirements)。HDE 申请无需包含科学有效的临床研究结果以证明设备能达到预期用途的有效性。然而,该申请必须包含足够的信息,以便 FDA 能够确定该医疗器械不会造成不合理或重大的疾病或伤害风险,并且综合考量现有医疗器械或替代医疗方案的潜在风险和效益,其可能带来的健康益处大于使用造成的伤害或疾病风险。此外,申请人必须证明目前尚无可用于治疗或诊断该疾病或病症的类似器械(Comparable devices),并且他们无法通过其他途径将该医疗器械推向市场。
经批准(Approved)的人道主义用途医疗器械 HUD 可获准上市销售。但该医疗器械仅可在获得机构审查委员会 IRB 批准用于 FDA 批准的适应症(Indication)后方可使用。HUD 的标签必须注明该医疗器械为人道主义用途医疗器械,并且尽管该医疗器械已获得联邦法律授权,但其针对特定适应症(Specific indication)的有效性尚未得到证实。
新分类申请 De Novo 数据库
De Novo 数据库(根据第 513(f)(2) 条对医疗器械进行的分类):https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/denovo.cfm
该数据库每周更新,可查看通过 De Novo 新分类申请已获准的新型医疗器械(New devices)列表。
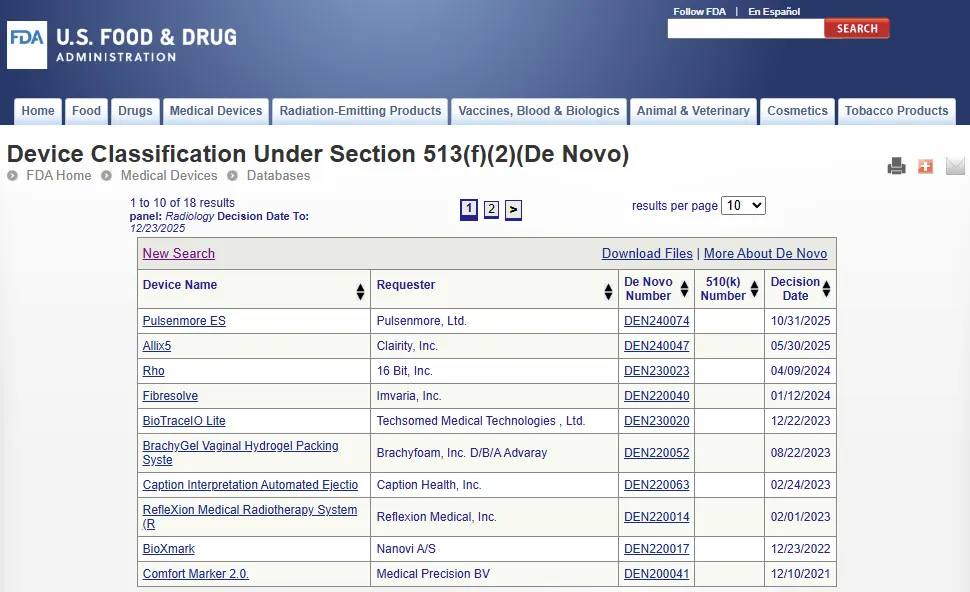
2012 年,《FDA 安全与创新法案》FDASIA 第 607 条(Section 607 of the Food and Drug Administration Safety and Innovation Act)对 FD&C 法案第 513(f)(2) 条(Section 513(f)(2) of the FD&C)进行了修订,为 De Novo 分类提供了第二种选择。在第二种途径中,如果申请方确定没有合法上市的器械(Legally marketed device)可以作为实质等同性判定的依据,则可以请求 FDA 根据该法案第 513(a)(1) 条(Section 513(a)(1))对该医疗器械进行基于风险的分类,而无需先提交 510(k)。
即对于低风险至中等风险的医疗器械,De Novo 分类有两种选择:
选项一:
任何收到针对 510(k) 申报的 NSE 决定的申请方都可以在收到 NSE 决定后的 30 天内提交 De Novo 申请,请求 FDA 对该器械进行基于风险的评估,将其归类为 I 类或 II 类医疗器械。 选项二:
任何确定没有合法上市的器械可供作为进行实质等效性判定依据的申请方,可以向 FDA 提交 De Novo 申请,请求 FDA 根据风险将该器械分类为 I 类或 II 类,而无需先提交 510(k) 申请并获得 NSE 判定。
通过 De Novo 新分类流程(De Novo process)获批的医疗器械可以上市销售,并可作为未来 510(k) 申报(510(k) submissions)的对比器械(Predicates)。
另外,需注意的是,自 2025 年 10 月 1 日起,所有 De Novo 申请的提交,除非获得豁免,否则必须使用 eSTAR 以电子方式提交。
510(k) 豁免医疗器械数据库
I 类和 II 类医疗器械 510(k) 豁免数据库:http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpcd/315.cfm
大多数 I 类器械(Class I devices)和部分 II 类器械(Class II devices)无需提交上市前通知 510(k)。
该数据库按器械类别列出了 510(k) 豁免(510(k) exempt)和良好生产规范 GMP / 质量体系豁免的器械列表。除非脚注(Footnote)进一步限定,否则此列表中的所有器械均为 510(k) 豁免器械。只有标有 (*) 的器械也免于 GMP,但一般记录保存要求和合规文件除外。
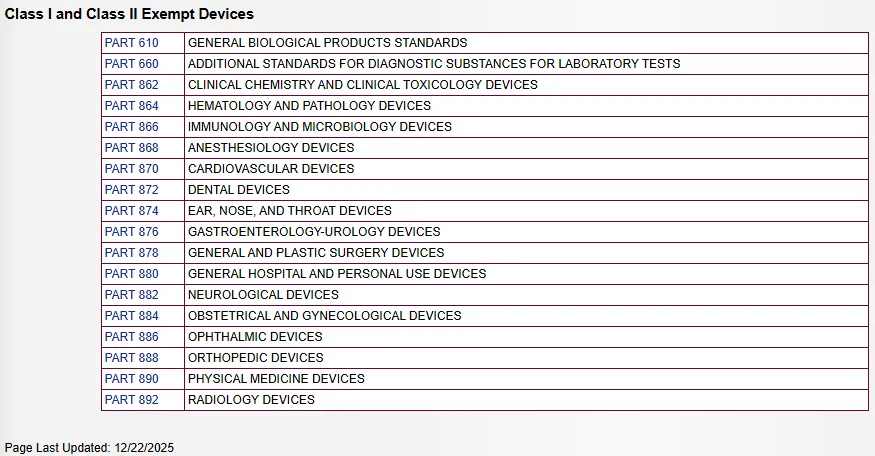
I 类医疗器械(Class I Devices):
FDA 已豁免几乎所有 I 类医疗器械(Class I devices)(保留器械除外)(Reserved Devices)的上市前通知(Premarket notification)要求,包括那些根据 1994 年 12 月 7 日和 1996 年 1 月 16 日《联邦公报》(Federal Registers)上发布的最终法规(Final regulation)中豁免的器械。注有“#\”的 510(k) 豁免具有某些限制,如脚注中所述。根据 21 CFR 第 862-892 部分(21 CFR Parts 862-892)确认豁免状态和任何适用的限制非常重要。器械豁免的限制在 21 CFR xxx.9 中有所规定,其中 xxx 指的是第 862-892 部分(Parts 862-892)。
如果制造商的器械属于 21 CFR 第 862-892 部分(21 CFR Parts 862-892)所定义的 I 类豁免器械的通用类别(Generic category),则在美国销售该器械前无需提交上市前通知申请(Premarket notification application)和获得 FDA 上市许可(FDA clearance)。但是,这些制造商必须注册其机构(Establishment)并列名器械通用类别(Generic category)或分类名称(Classification name)。注册和列名信息(Registration and listing information)可通过 FDA 的统一注册和列名系统 FURLS(FDA’s Unified Registration and Listing System)的医疗器械注册和列名模块 DRLM(Device Registration and Listing Module)提交。
II 类医疗器械(Class II Devices):
FDA 还公布了一份 II 类(特殊管制要求)(Special controls)器械列表(这些器械标注为“(II)”),根据 1997 年《 FDA 现代化法案》(FDAMA)(Food and Drug Administration Modernization Act of 1997)或2016 年《21 世纪治愈法案》(Cures Act)(21st Century Cures Act of 2016),这些器械在某些限制条件下可豁免于上市前通知要求(Exempt from premarket notification requirements)。FDA 认为,这些豁免将使制造商无需为这些医疗器械提交上市前通知申报(Premarket notification submissions),并使 FDA 能够将用于审查此类申报的资源重新分配到更重要的公共卫生问题上。FDA 采取此行动是为了满足 FDAMA 和 Cures Act 法案的要求。II 类器械标注有“(II)”。需注意的是,II 类器械不免于 GMP 要求。
以上便是 FDA 医疗器械相关的一些公共查询数据库的介绍,FDA 下属的医疗器械和放射健康中心 CDRH(Center for Devices and Radiological Health)还提供许多其他与医疗器械(Medical devices)和发射辐射产品(Radiation-emitting products)相关的数据库,如需获取 CDRH 数据库中未包含的信息,可以根据《信息自由法案》(Freedom of Information)提出申请。












