



重磅!美国疾控中心紧急宣布接受N95口罩中国标准!欧盟也同步开启口罩绿色通道!

 17600
17600近期,欧美口罩的认证和标准成为广大外贸出口和货代物流企业关注的焦点!据搜航网最新获悉,上周,美国疾病控制与预防中心(CDC)正式发布涉及口罩标准规定的《优化N95口罩供应策略:危机/替代策略》!
CDC宣布接受N95口罩中国标准

这是一个事关所有计划出口美国的外贸物流企业的事件,公告指出:新冠肺炎疫情期间,当N95口罩供给不足时,按下表中标准生产的口罩,是N95口罩合适的替代品。其中包括采用中国标准GB 2626-2006 和GB/T 18664—2002生产的口罩。


Strategies for Optimizing the Supply of N95 Respirators: Crisis/Alternate Strategies
Crisis/Alternate Strategies
These crisis capacity or alternate strategies accompany and build on the conventional and contingency capacity strategies. The following measures are not commensurate with current U.S. standards of care.
However, individual measures or a combination of these measures may need to be considered during periods of expected or known N95 respirator shortages. It is important to consult with entities that include some combination of: local healthcare coalitions, federal, state, or local public health officials, appropriate state agencies that are managing the overall emergency response related to COVID-19, and state crisis standards of care committees.
Even when state/local healthcare coalitions or public health authorities can shift resources between health care facilities, these strategies may still be necessary.

目前在疫情期间,进入美国市场都需要进口商向FDA申请获取口罩的紧急使用资质,符合以下标准的N95口罩,可以简化原美国FDA注册流程以及NIOSH的检测流程,快速投入使用。
国家 | 执行标准 | 可接受的产品等级 | 标准/指导文件 | 防护系数≥ 10 |
澳大利亚 | AS/NZS 1716:2012 | P3 P2 | AS/NZS 1715:2009 | YES |
巴西 | ABNT/NBR 13698:2011 | PFF3 PFF2 | Fundacentro CDU 614.894 | YES |
中国 | GB 2626-2006 | KN100 KP100 KN95 KP95 | GB/T 18664—2002 | YES |
欧盟 | EN 149-2001 | FFP3 FFP2 | EN 529:2005 | YES |
日本 | JMHLW-2000 | DS/DL3 DS/DL2 | JIS T8150: 2006 | YES |
韩国 | KMOEL-2017-64 | Special 1st | KOSHA GUIDE H-82-2015 | YES |
墨西哥 | NOM-116-2009 | N100, P100, R100,N99, P99, R99, N95, P95, R95 | NOM-116 | YES |
美国NIOSH要求 | NIOSH approved | N100, P100, R100,N99, P99, R99, N95, P95, R95 | OSHA 29CFR1910.134 | YES |
早前,美国副总统彭斯在3月5日新闻发布会上告诉大家,“除非病了,否则无需购买口罩”。

欧盟开启口罩绿色通道
不仅美国,同样深陷新冠肺炎疫情的欧洲,同样特事特办,紧急开通了口罩类防疫物资的绿色通道。
近日,欧美紧急放宽口罩等防疫物资准入要求(CE认证和FDA认证),需要机构认证的产品在完成合规性评估程序之前(即取得CE/FDA标志之前)可以先出口,但是要确保认证工作会继续完成。
欧盟成员国主管当局可在疫情期间评估和集中采购没有CE标记的防疫产品,该产品仅可以提供给医护人员使用,不能在市场上流通销售。如果你的产品不是政府集中采购,且要在当地市场上销售的,则不属于上述放宽准入的条件范围。
2020年3月13日,欧盟会员会在欧洲官方杂志( Official Journal of the EuropeanUnion)发布了疫情期间针对医疗器械和个人防护用品 (PPE)的符合性评价和市场监督程序的建议。

医疗器械方面:
◆如果市场监督机构确定产品符合医疗器械的基本安全和性能要求,即使其符合性评价还未完成,市场监督机构可以允许其在一定的时间内进行销售,同时该产品必须继续完成其符合性评价过程。
◆成员国主管当局也可在疫情期间评估和组织采购没有CE标记的医疗器械,该产品仅可提供给医疗工作者使用,不能在市场上流通销售。同时市场抽查将会重点抽查防疫相关医疗器械,以防止不合格产品导致严重风险。
个人防护用品(PPE)方面:
◆涉及的产品包括抛弃式和可重复使用的口罩、可重复使用的工作服、手套和眼罩等(主要是预防病毒和有害物质的产品)。需要具有PPE法规授权资格的公告机构进行符合性评价。
◆应急审批产品如果不采用PPE法规协调标准作为产品技术要求而采用其它技术要求,比如WHO的推荐要求,须确保采用的技术要求与PPE法规基本健康与安全要求同等防护水平。公告机构对这类采用其它技术要求的PPE产品进行发证时,需要立即通知主管当局和其它PPE法规的公告机构。
◆如果市场监督机构确定产品符合PPE法规的基本健康和安全要求,即使其符合性评价还未完成,市场监督机构可以允许其在一定的时间内进行销售,同时该产品必须继续完成其符合性评价过程。
◆成员国主管当局也可在疫情期间评估和组织采购没有CE标记的PPE产品,该产品仅可提供给医疗工作者使用,不能在市场上流通销售。同时市场抽查将会重点抽查防疫相关PPE产品,以防止不合格产品导致严重风险。
也就是说,只要处于正在进行符合性评估的过程中,就可以在没有CE标志的情况下先行进入欧盟市场。由市场监督部门进行抽查,发现问题再进行处罚。
重点如下!!!
◆成员国可采购安全有效,但没有CE标记的医疗产品;
◆紧急物资专供医疗人员使用,不可在市场上流通;
◆仅疫情期间有效。
关于CE标识
CE标签就像一把巨伞,底下是规定各类产品安全标准、细分到不同材料和生产模式等的各种欧盟指令。自1985年成立以来,它就成为了高质量、高标准和严格执法的标志,缺少这一标志的商品将不予获准进入欧盟市场。
如今CE标识已经成为了全球认可的质量标志,CE标志可以证明该批在欧盟制作或进口至欧盟成员国的产品符合质量标准,满足保护消费者健康、供应链安全和环境可持续发展的要求。
| 欧盟口罩要求 |
|---|
CE认证是欧盟实行的强制性产品安全认证制度,目的是为了保障欧盟国家人民的生命财产安全。
|
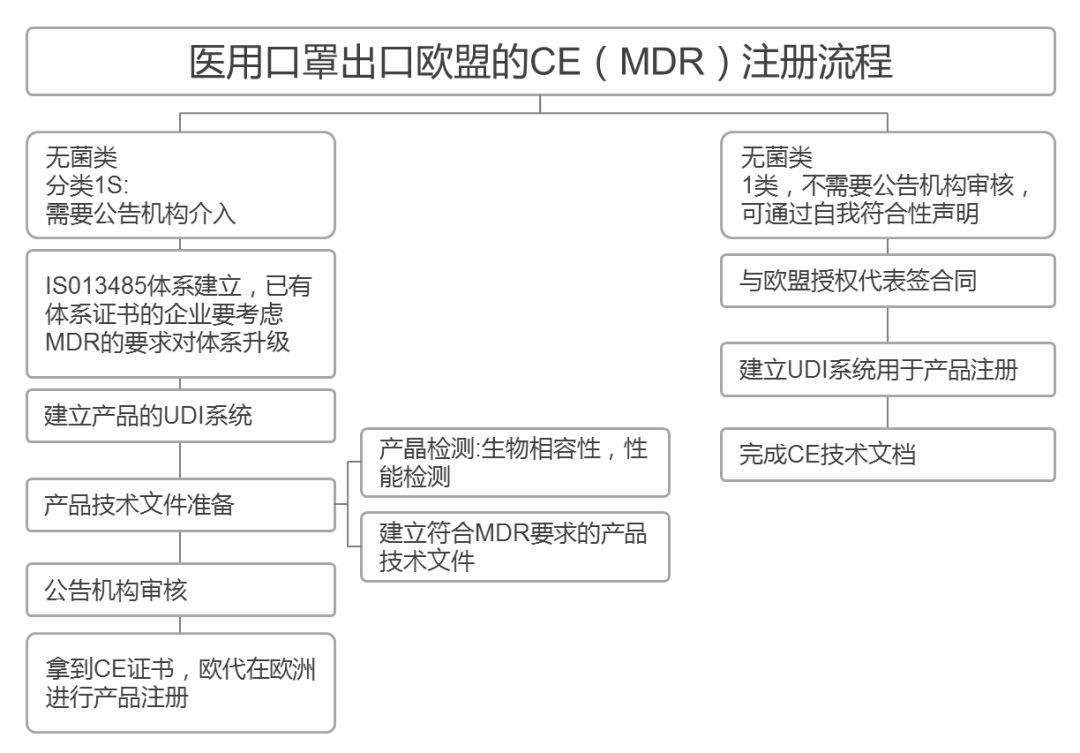

新手小白可以问自己的发证机构两个问题:
01、贵司是否为NB机构? 机构号是否可以查询?
02、出具的CE证书在官网可查吗?

NB机构可以理解为被欧盟授权或认可的机构。如果CE证书是NB机构发证的,在欧盟就具有一定的效应,清关的风险才会相对较小。
正常情况下,根据欧盟法规,所有出口欧盟的产品都需要获得CE认证,加贴CE标识才能进入欧洲市场。CE认证的审核和发证,欧盟公布了一系列由欧盟统一监管和认证资质授权的机构,并授予每家机构一个唯一的四位数编码即公告号,CE证书的申请和颁发就由对应法规和指令授权的公告号机构颁发。
在欧盟官方网站-欧盟公告机构查询官网,厂家可以查询到目前从0001-2786 两千多家欧盟公告号机构详细信息,每家机构对应的指令和法规授权以及发证机构信息都可在该网站查询到。
附:CE认证查验:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=notifiedbody.main
附:FDA查验(出口美国需要FDA和NIOSH):
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfRL/rl.cfm

近期,大家都在对口罩等医疗物资的欧盟标准犯愁,都是一知半解的状态。在此附上原文查看:
欧盟成员国名单(27国):
奥地利、比利时、保加利亚、塞浦路斯、捷克、克罗地亚、丹麦、爱沙尼亚、芬兰、法国、德国、希腊、匈牙利、爱尔兰、意大利、拉脱维亚、罗马尼亚、立陶宛、卢森堡、马耳他、荷兰、波兰、葡萄牙、斯洛伐克、斯洛文尼亚、西班牙、瑞典。
(文章来源@搜航网)










