




【美国进口清关】FDA 医疗器械:机构注册&器械列名

 2521
2521

一键完成百千笔付款,超低费率+极速到账,一年轻松帮你省下数十万。
机构注册和器械列名(Registration and listing)是适用于所有医疗器械的一般管制要求(General control)。医疗器械机构(Medical device establishments)必须向 FDA 注册并列名在其机构内生产(Manufactured)、制备(Prepared)、培养(Propagated)、配制(Compounded)、组装(Assembled)或加工(Processed)的医疗器械。所有注册和列名信息必须以电子方式提交,除非获得豁免。据 FDA 介绍,注册和列名使美国食品药品监督管理局 FDA 能够了解医疗器械机构的所在地及其生产的医疗器械信息。完成初始注册(Initial registration)后,后续注册将被视为年度注册和列名(Annual registration and listing)。必须在每年 10 月 1 日至 12 月 31 日期间提交年度注册(Annual registration)。所有需要注册的机构(Establishments)必须首先访问 FDA 器械设施用户费用网站 DFUF(Device Facility User Fee)缴纳用户费用。收到支付识别码 PIN 和支付确认码 PCN 后,即可前往 FDA 的 FURLS 网站完成机构注册和器械列名流程。
机构注册 & 器械列名概述
根据美国《联邦食品、药品和化妆品法案》(FD&C Act)及《公共卫生服务法案》(PHS Act),FDA 拥有对进口或拟进口至美国的食品、化妆品、药品、生物制品、医疗器械及烟草制品实施监管的法定权限(Legal authority)(《联邦食品、化妆品和化妆品法案》第 701 和 801 条,《公共卫生服务法案》第 351 条)(Sections 701 and 801 of the FD&C Act;section 351 of the PHS Act)。FDA 还拥有监管发射辐射的电子产品(Radiation-emitting electronic products)进口的法定权限(《联邦食品、药品和化妆品法》第 536 条)(Section 536 of the FD&C Act)。
凡参与生产和分销拟在美国使用的医疗器械(Medical devices)的营业场所(也称为机构或设施)(Establishments or facilities)的所有者或经营者(Owners or operators),必须每年向美国食品药品监督管理局 FDA 注册。此流程称为机构注册(Establishment registration)。商业分销(Commercial distribution)指任何旨在供人类使用的医疗器械分销行为,该医疗器械被持有或提供出售,完整定义参见《联邦法规汇编》第 21 编第 807.3(b) 条(21 Code of Regulations, CFR, 807.3(b))。
大多数需要注册的机构(Establishments)还必须列名其所生产的医疗器械以及在该机构内开展的与这些医疗器械相关的活动(Title 21 CFR Part 807)。如果器械在美国上市前需要提交上市前申报(FDA premarket submission),则其所有者/经营者(Owner/operator)还应提供 FDA 上市前申报编号(FDA premarket submission number)(510(k)、De Novo、PMA、PDP、HDE)。
医疗器械列名所需信息包括:
1,机构注册编号和名称。
2,在机构内开展的活动。示例:制造(Manufacturing)、贴标(Labeling)。
3,监管信息(Regulatory Information):
产品代码(Product Code):如果你的医疗器械豁免于上市前提交(Exempt from premarket submission)或在 1976 年 5 月 28 日前已上市(修正案前医疗器械)(Pre-Amendments)。 上市前提交编号(Premarket submission number):如果你的医疗器械未获豁免或属于修正案后医疗器械(Pre-Amendments)。 示例:K239999;P239999;DEN239999;H239999。
自 2002 年以来,CDRH 开始实施医疗器械用户费用计划(Medical device user fee program),要求受监管的医疗器械行业(Device industry)向 CDRH 缴纳用户费用(User fees),以促进监管决策效率。美国国会已授权 FDA 向医疗器械企业征收年度企业注册费(Annual establishment registration)。所有需要注册的机构现在都必须按照 FDA《安全与创新法案》FDASIA(Food and Drug Administration Safety and Innovation Act)的要求缴纳年度注册用户费(Annual registration user fee)。
2025 财年,小型企业不享有 MDUFA 机构注册费(Establishment registration fee)豁免或减免(Waivers or reductions)。2026 财年,FDA 可能会对通过小型企业认定计划 SBD(Small Business Determination)认证且经 FDA 认定缴纳该费用将造成财务困难的某些小型企业免除年度机构注册费(不包括初始注册费)。
FDA 用户费用(User fee)金额通常每年都会有所调整,并由法规规定(Statute)。用户费用(User fee)周期以财政年度为单位(Fiscal year),即从当年 10 月 1 日至次年 9 月 30 日。
2025 财年和 2026 财年的年度注册用户费用(Annual registration user fee)如下:
2025 财年(2024 年 10 月 1 日 - 2025 年 9 月 30 日):$9,280 2026 财年( 2025 年 10 月 1 日 - 2026 年 9 月 30 日):$11,423
所有需要注册的医疗器械机构(Device establishments)必须在每个财政年度(Fiscal Year)的 10 月 1 日至 12 月 31 日之间完成年度注册(Annual registration)。
自 2012 年 10 月 1 日起,所有需要注册的机构(Establishments)必须在完成注册前支付用户费用(User fee)。费用通过 FDA 的器械设施用户费用网站 DFUD(Device Facility User Fee)进行付款:https://userfees.fda.gov/OA_HTML/furls.jsp。年度注册用户费用后(Annual registration user fee)完成支付后,FDA 将通过电子邮件向你发送支付识别码 PIN(Payment identification number)。付款处理完毕后,一封包含支付确认码 PCN(Payment confirmation number)的电子邮件将发送到你账户关联的电子邮件地址。收到 PIN 和 PCN 后,就可以通过 FDA 的电子注册和列名系统(Electronic registration and listing system)FURLS:https://www.access.fda.gov/oaa/ 进行机构注册和器械列名。
必须同时提供 PIN 和 PCN 才能完成 FURLS 注册。如果在都收到 PIN 和 PCN 之前提前开始操作,将无法在 FURLS 系统保存已填写的任何信息,且后续重新登录系统时需重新输入所有内容。
完成机构注册及器械列名后,在美国进口清关过程中,当 Entry 申报信息提交至 FDA 时,FDA 会将提交的信息与 CDRH 的机构注册和器械列名数据库进行比对,以核实申报的制造商(Manufacturer)和托运人(Shipper)是否已在 FDA 注册。同时 FDA 还会使用 CDRH 的数据系统来核实申报的进口商是否已注册。申报产品的列名信息还通过将申报的产品描述与 CDRH 的机构注册和产品列名数据库进行比对来验证。
如果提交的信息与 CDRH 机构注册和器械列名数据库相符,则视为合规性得到验证;如果信息不符,FDA 可能会收集更多信息或扣留产品(Detain)。如果企业(Firm)未完成必要的机构注册和器械列名,则其产品将被拒绝入境(Refusal)。
通过 FDA 的医疗器械机构注册和产品列名数据库,可以查询任何已在 FDA 注册的医疗器械机构的注册信息。在适用情况下,该数据库亦包含已在 FDA 进行列名的医疗器械。
谁必须注册、列名并缴纳费用
凡从事生产和分销拟在美国使用的医疗器械(Medical devices)的营业场所(也称为机构或设施)(Establishments or facilities)的所有者或经营者,必须每年向 FDA 注册。此过程称为机构注册(Establishment registration)。大多数需要注册的机构(Establishments)还必须列名其所持有的医疗器械以及在这些器械上进行的活动(Title 21 CFR Part 807))。
下表根据机构开展的活动类型,详细列出了注册和列名的要求。图表中还有一栏,列明哪些类型的活动需要缴纳机构注册费(Establishment registration fee)。
国内机构(位于美国境内)(Domestic Establishments)
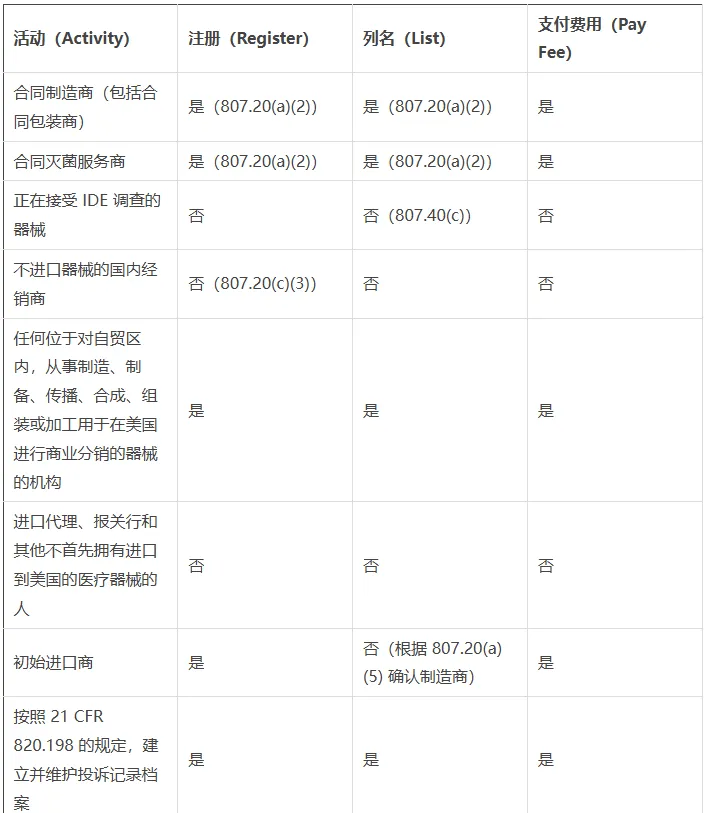
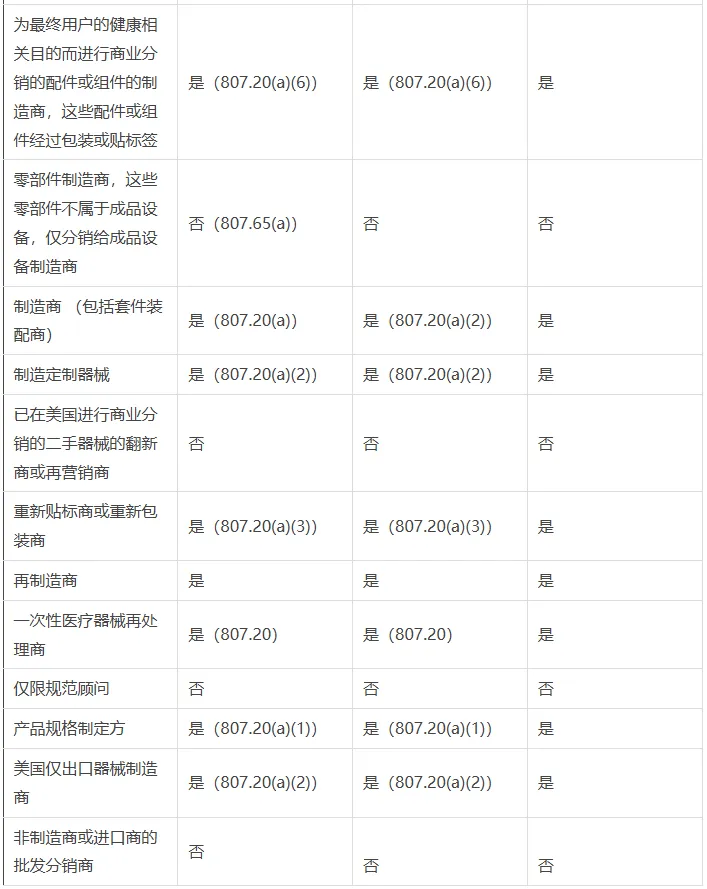
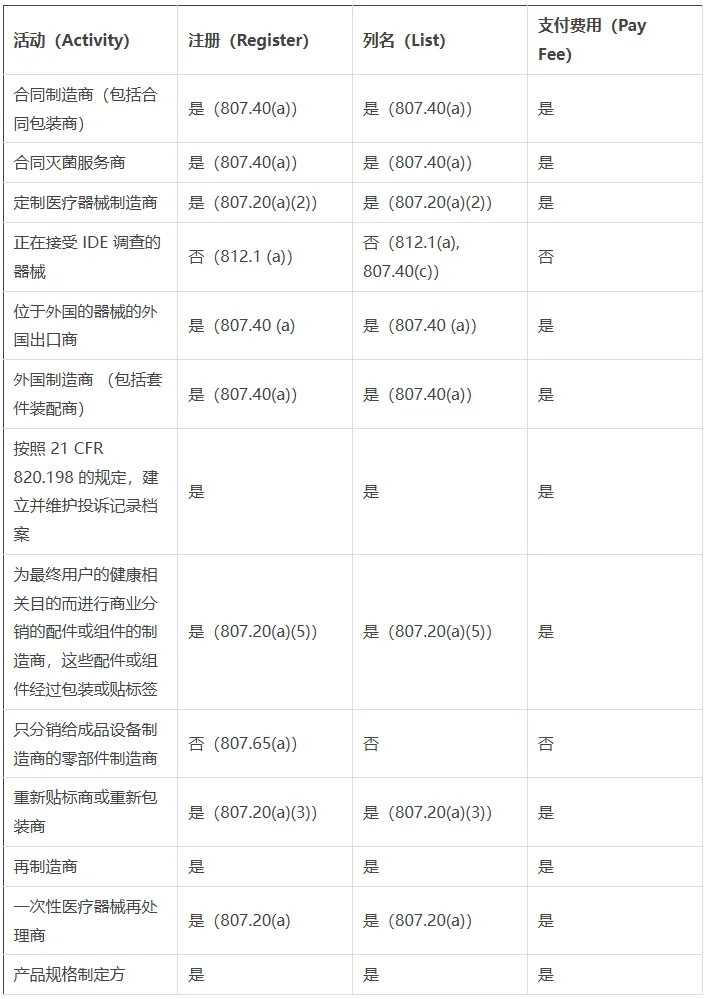
合同制造商(Contract Manufacturer):
按照其他机构的规格制造成品医疗器械。 合同灭菌服务商(Contract Sterilizer):
为其他机构的器械提供消毒灭菌服务。 外国出口商(Foreign Exporter):
向美国出口或提供出口在外国制造、制备、传播、合成或加工的器械,包括最初在美国制造的器械。外国出口商必须在美国境外拥有营业地址。 初始进口商(Initial Importer):
任何进口商,指将外国制造商生产的器械进一步营销给最终交付或销售给最终消费者或用户,但未重新包装或以其他方式更改器械或器械包装的容器、包装材料或标签的进口商。 制造商(Manufacturer):
通过化学、物理、生物或其他程序制造任何符合《联邦食品、药品和化妆品 (FD&C) 法案》第 201(h) 条(Section 201(h) of the Federal Food, Drug, and Cosmetic (FD&C) Act)中“医疗器械”(Device)定义的物品(Article)。 重新包装商(Repackager):
将散装成品医疗器械包装或将制造商生产的器械重新包装到不同的容器中(不包括运输容器)。 重新贴标商(Relabeler):
更改原制造商提供的标签内容,以企业自己的名义销售。重新贴标商不包括那些不更改原标签而仅添加自己名称的机构(Establishments)。 再制造商(Remanufacturer):
任何对成品医疗器械进行加工、调节、翻新、重新包装、修复或进行任何其他行为,以显著改变成品器械的性能或安全规格或预期用途的相关方。 一次性医疗器械再处理商(Reprocessor of Single Use Device):
对一次性使用器械执行再制造操作。 产品规格制定方(Specification Developer):
为以企业自身名义分销但不进行生产的器械制定规范。这包括除制定规范外,还安排合同制造商生产贴有其他机构名称器械的机构(Establishment)。 美国仅出口器械制造商(U. S. manufacturer of export only devices):
生产不在美国销售且仅供出口到国外的医疗器械。
常见的一些免于注册的机构(Establishments)
1,零部件制造商(Component Manufacturer)
提供用于医疗器械制造或组装(Manufacture or assembly)的原材料或部件(Raw materials or components)(尚未准备好使用) 仅提供给成品医疗器械制造商(Finished device manufacturer)
2,仅用于兽医用途的医疗器械制造商
FDA 兽医药品中心 CVM(Center for Veterinary Medicine)负责监管仅供兽医用途的医疗器械
3,持牌执业医师(Licensed Practitioner)
制造或以其他方式改造仅供自身诊疗使用的医疗器械 不得将医疗器械分发(Distribute)给其他从业人员(Practitioners)
4,安装程序(Installer)
由制造商代理提供(By manufacturer’s agent) 由用户提供(By user)
5,美国国内经销商(Domestic Distributor)
6,零售店(Retail Establishment)
直接向最终用户(End users)提供医疗器械 包括药店(Pharmacies)和外科用品店(Surgical supply outlets) 注意:如果器械来自国外机构(Foreign establishments),需确保产品按要求贴上标签
7,仅限研究用途的医疗器械制造商(Manufacturer of research use only devices)
按要求标记医疗器械并仅用于研究、教学或分析 尚未投入商业流通(Not introduced into commercial distribution) 仅适用于美国国内机构(Only applies to domestic establishment)
何时注册和列名
年度注册(Annual Registration)
即使注册信息未发生任何变更,也必须每年在 10 月 1 日至 12 月 31 日期间进行年度注册。
每年 10 月 1 日至 12 月 31 日期间,必须在审核注册信息的同时,同步审核列名信息。如有任何更新,请在此期间提交。
初始注册(Initial Registration)
国内机构(Domestic establishment)(位于美国境内)必须在开展业务活动或将器械投入商业分销后 30 天内提交注册和/或列名信息(Listing information)。外国机构(Foreign establishments)(位于美国境外)必须在首次将医疗器械出口到美国之前进行注册,美国国内进口商在进口产品前必须进行注册。另外,对于初始进口商(Initial Importer),必须在首次将医疗器械进口到美国之前需要进行机构注册(仅需注册,无需列名医疗器械),且需标识每台进口医疗器械的制造商。
请注意,如果你的产品需要上市前通知许可或批准(Premarket notification clearance or approval),则必须等到上市前申报(Premarket submission)(例如 510(k)、PMA 等)获得许可(Cleared)或批准(Approved)后,才能注册你的机构并进行器械列名(List the device)。若未完成相关流程,FDA 将与 CBP 合作扣留并扣押货物(Detain and hold shipment)。
更新注册和列名信息(Update Registration & Listing Information)
机构所有者(Owners)或经营者(Operators)均可随时通过 FURLS 系统以更新其注册和列名信息中的变更内容。列名信息变更(Changes to listings)示例包括:
另一款器械(Device)被引入商业流通(Commercial distribution); 之前列名(Listed)的器械(Device)发生变更(例如其生产地变更); 之前列名的器械停止商业销售或恢复商业销售。
企业注册(Establishment registrations)基于 FDA 的财政年度(Fiscal year),财政年度从 10 月 1 日到 9 月 30 日。FDA 将继续把企业的注册状态视为有效(Active),直至每个日历年度结束。
如何注册和列名
一般信息(General Information)
2007 年《FDA 修正法案》(FDAAA)(The Food and Drug Administration Amendments Act of 2007)规定,除非 FDA 授予豁免(Grants a waiver),否则所有注册和列名信息(年度、初始或更新)都必须以电子方式提交。
医疗器械机构(Eestablishment)注册分为两个步骤。首先,必须缴纳年度用户注册费(Annual registration user fee)。缴费完成后,即可进行注册流程。注册过程需完成以下步骤:
1,已支付年度用户注册费
2,已通过电子方式提交了注册和列名信息,且
3,并收到了 FDA 的电子邮件通知,确认所有要求均已满足。
支付年度注册费(Paying the Annual Registration Fee)
通过 FDA 的器械设施用户费用 DFUF(Device Facility User Fee)网站上以电子方式支付年度注册费(Annual registration fee)。
在 DFUF 网站上完成支付后,将收到你的支付识别码(PIN)。当 FDA 已成功收到你的付款,你将收到一封包含支付确认信息和获取支付确认码 PCN 说明的电子邮件。此过程可能需要数天时间,因此 FDA 建议务必在注册前至少提前几天完成支付。
收到付款确认后,即可继续进行设施(Facility)注册。
注册你的设施(Registering Your Facility)
注册和列名信息(Registration and listing information)通过 FDA 统一注册和列名系统 FURLS(Unified Registration and Listing System)的器械注册和列名模块 DRLM(Device Registration and Listing Module)提交。
FURLS 是 FDA 用于机构注册和器械列名的电子系统。FURLS 有两种账户类型:所有者/经营者账户 OO(Owner/operator)和官方通讯员账户 OC(Official correspondent)。OO 账户(Owner/operator account)通常被称为 “企业账户”(Enterprise)或“主账户”(Primary account),OO 账户分配给直接负责机构注册活动的企业或所有者(Corporation or proprietor)。OO 账户可以创建新的 FURLS 子账户及 OC 账户(Official correspondent accounts),以及可以创建、更新及注销(Deactivate)注册机构和列名器械。而 OC 账户是由所有者/经营者 OO 账户(Owner/operator account)创建的账户,OC 账户可以为其负责的任何机构填写注册和列名信息(Registration and listing information)。
年度注册(Annual Registration)
1,在器械设施用户费用网站 DFUF(Device Facility User Fee website)完成付款并获取支付识别码 (PIN) 和支付确认码 (PCN) ;
2,使用你的 FURLS 账户 ID 和密码登录 FURLS ( https://www.access.fda.gov/oaa/)。 如果你正在进行年度注册,你已经拥有账户 ID 和密码,请勿创建新账户。创建新账户将导致你无法访问当前注册信息;
3,选择 DRLM 按钮(器械注册和列名模块)(Device Registration and Listing Module);
4,从 DRLM 主菜单中选择“年度注册”(Annual Registration)链接。必须选择“年度注册”(Annual Registration)链接并完成此流程,你的机构(Establishment)才会被视为已在本财政年度完成注册。点击“年度注册”(Annual Registration)链接后,你还可以更新你的注册和/或列名信息(Registration and/or listing information);
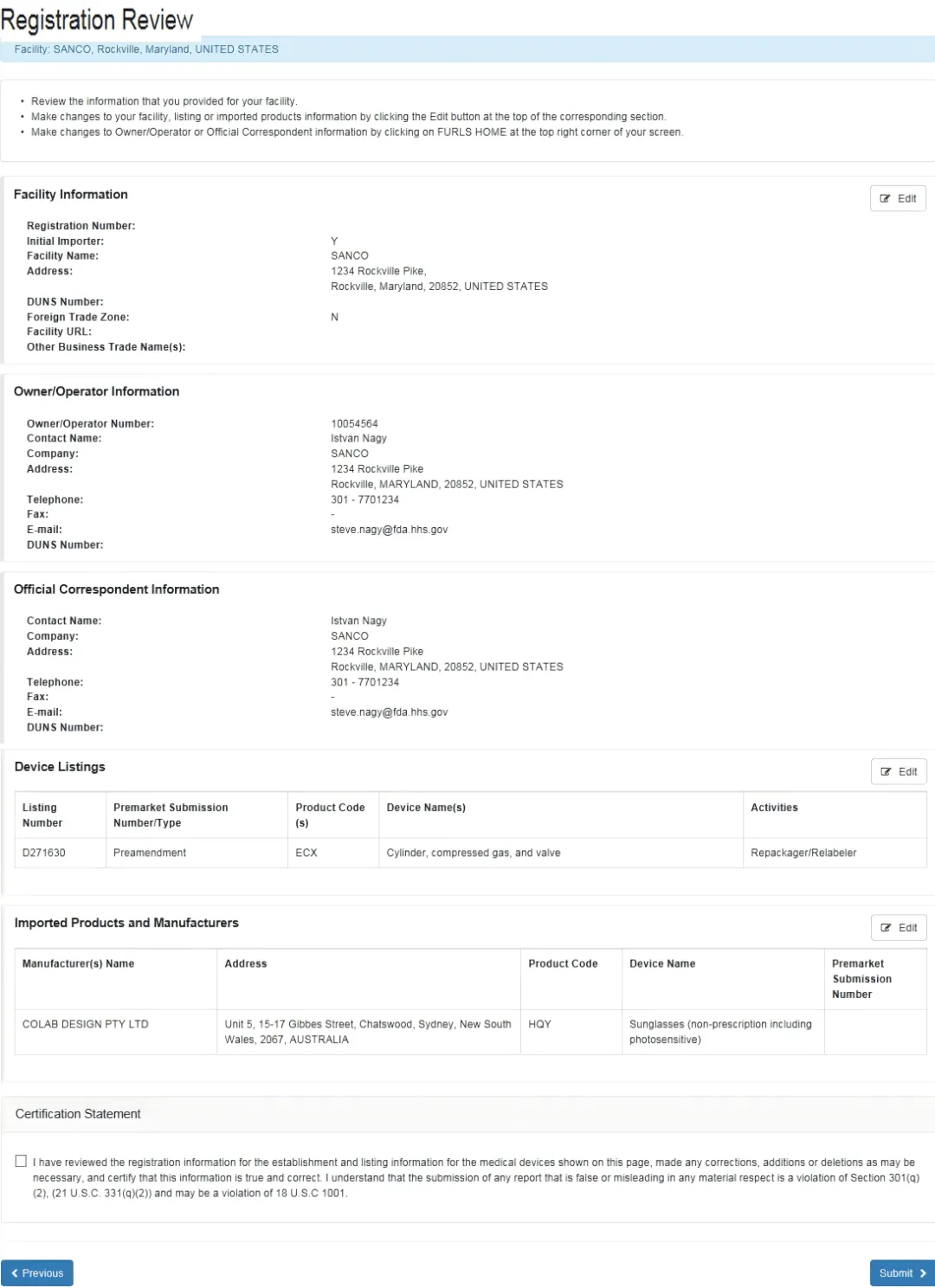
5,核对(Review)你的机构(Establishment)的注册信息并进行更新(如有必要);
6,核对(Review)你的器械的列名信息(Listing information)并根据需要进行更新。如果你是初次进口商(Initial importer),请核对你进口器械的制造商列表(List of manufacturers)。如果你是外国机构(Foreign establishment),请核对你出口的每种器械已知的进口商列表。如果此信息此前未被录入,则必须在年度注册期间完成录入,才能完成本财年度的注册流程;
7,确认所有信息无误后,点击“提交”(Submit)按钮;
8,当系统出现提示时(When prompted),请输入你从 FDA 财务管理办公室(Office of Financial Management)收到的用于支付机构注册费(Establishment registration fee)的 PIN 码和 PNC 码。必须输入这些信息,FDA 才能接受你的注册。如果你没有收到输入 PIN 码/PCN 码的提示,请发送电子邮件至 reglist@cdrh.fda.gov。未输入这些识别码,你的注册将无法完成。一旦收到确认页面(Confirmation screen),FDA 将视你已完成注册;
9,如果你需要更改你的列名信息,请返回主菜单并选择“更改”(Change)、“取消”(Cancel)或“重新激活列名信息”(Reactivate Listings)以更新你的列名信息。
初始注册(Initial Registration)
1,在器械设施用户费用网站上 DFUF(Device Facility User Fee website)完成付款并获取支付识别码 PIN 和支付确认码 PCN;
2,访问 FURLS:https://www.access.fda.gov/oaa/;
3,如果你之前从未注册过器械机构(Device establishment),则需要先为所有者/经营者(Owner/operator)创建一个 FURLS 帐户。请注意:如果你已经拥有该所有者/经营者(Owner/operator)的帐户,则必须使用该用户 ID 和密码登录 FURLS;
4,设置好 FURLS 帐户 ID 和密码后,选择 DRLM 按钮(器械注册和列名模块)(Device Registration and Listing Module);
5,从 DRLM 主菜单中选择“注册医疗器械设施”(Register a Medical Device Facility)链接;
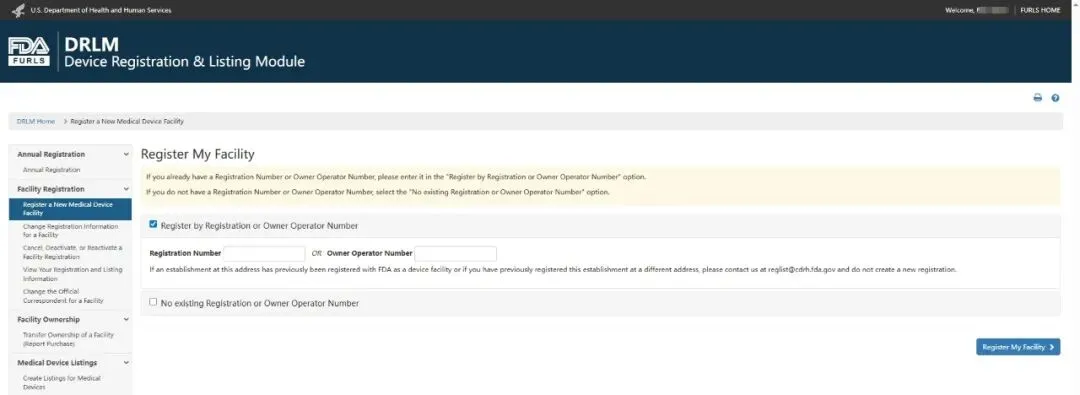
6,如果你没有任何现有注册,你将看到一个页面,要求你确认你的机构(Establishment)现有的所有者/经营者(Owner/operator)编号或注册编号。请将方框留空,并选择“无现有注册或有者/经营者编号”(No existing registration or OO number);
7,如果你已有注册,系统会要求你确认正在注册的机构是否未出现在显示的列表(List)中。如果该机构已注册,请勿创建重复记录。如果该机构未出现在显示的列表(List)中,请选择“注册新设施”(Register a New Facility);
8,你在创建或更新 FURLS 帐户时输入的所有者/运营者(Owner/Operator)和官方通讯员信息(Official Correspondent information)将显示在此处。如需更改所有者/运营者或官方通讯员信息,你需要退出 FURLS 的 DRLM 部分并返回帐户管理(Account Management)。如果信息正确,请选择“继续注册”(Continue Registration);
9,输入你设施(Facility)的必要信息,然后选择“继续注册”(Continue Registration);
10,系统将提示你输入你制造(Manufacture)、加工(Process)、分销(Distribute)或进口(Import)的器械的详细信息。制造商(Manufacturers)、加工商(Processors)和分销商(Distributors)必须列名(List)在每个设施中生产(Produced)或加工(Processed)的所有器械。初始进口商(Initial importers)(将医疗器械从国外制造商引入美国,并且首次将该器械分销到美国市场的美国境内机构或个人)必须列名其进口的所有器械的制造商(Manufacturers);
A,对于除初始进口商以外的所有设施(Facilities):
1),列名所有在此设施内生产或加工(Produced or processed)的器械。
2),对于每项列名产品(For each listing),请确定你的产品是否需要上市前通知/批准(Premarket notification/approval),或是否可豁免。请注意:如果某一器械(Device)需要上市前通知许可或批准(Premarket notification clearance or approval),则只能在上市前申请(510(k), PMA, PDP, HDE)获得许可或批准(Cleared or approved)后才能进行器械列名(Listed)。如果这是你唯一需要注册列名的器械,则请在上市前申报获得许可或批准(Premarket submission is cleared or approved)后才注册你的机构(Establishment)。
3),如果你的上市前申报已获许可或批准(Premarket submission is cleared or approved),你需要完成以下步骤以列名你的器械(List your device):
输入上市前申报编号(Premarket submission number); 输入专有名称(Proprietary name);即你的医疗器械在美国市场销售时使用的所有专有名称(Proprietary names)。你可将此信息标记为“Confidential”机密,这样该名称就不会公开显示在 FDA 的公开注册和列名数据库中(Public registration and listing database)。 确认(Identify)你在器械上或对器械执行的操作。
4),如果你的器械豁免于上市前通知/批准(Exempt from premarket notification/approval),你将需要执行以下操作:
获取产品代码(Product code); 将上市前申报编号(Premarket submission number)留空; 在筛选框(Filter box)中输入产品代码,然后点击“筛选”(Filter); 选中产品代码旁边的单选按钮(Radio button),然后点击“继续”(Continue); 确认(Identify)你在器械上或对器械执行的操作; 输入专有名称(Proprietary name)。
B,对于初始进口商(在医疗器械进口至美国后,首次取得其所有权并投入商业流通的机构)
1),在“识别制造商”(Identify Manufacturers)页面上,点击“搜索并添加产品”(Search & Add Products)按钮;
2),使用器械列名编号(Device listing number)、企业注册编号(Establishment registration number)或企业名称和地址(Establishment name and address)来识别制造商;
3),在下一页中,在与你的器械制造商相匹配的注册信息下,选择你要导入的器械,然后单击“添加选定产品”(Add Selected Products)按钮。
11,确认所有信息正确无误后,点击“提交”(Submit)按钮;
12,当系统出现提示时(When prompted),请输入你从财务管理办公室(Office of Financial Management)收到的用于支付机构注册费(Establishment registration fee)的 PIN 码和 PNC 码。必须输入这些信息,FDA 才能接受你的注册。如果你没有收到输入 PIN 码/PCN 码的提示,请发送电子邮件至 reglist@cdrh.fda.gov。未输入这些识别码,你的注册将无法完成。一旦收到确认页面(Confirmation screen),FDA 将视你已完成注册。
13,如果你需要更改你的列名信息,请返回主菜单并选择“更改”(Change)、“取消”(Cancel)或“重新激活列名信息”(Reactivate Listings)以更新你的列名信息。
美国代理(U.S. Agents)
任何从事制造(Manufacture)、制备(Preparation)、扩增或培养(Propagation)、配制(Compounding)或加工(Processing)进口到美国的医疗器械的外国机构(Foreign establishment)都必须为该机构指定一名美国代理(U.S. agent)。
外国机构的美国代理信息需通过 FDA 统一注册和列名系统(FDA Unified Registration and Listing System)(FURLS 系统)以电子方式提交,这是机构注册流程的一部分。每家外国机构只能指定一名美国代理。外国机构可以(但并非必须)将其美国代理指定为其官方通讯员(Official correspondent)。外国机构(Foreign establishment)应提供美国代理的姓名、地址、电话号码、传真号码和电子邮件地址。
指定的美国代理需完成一个自动化流程,以确认其同意担任该机构的美国代理。该自动化流程将向美国代理发送电子邮件验证请求。相应的美国代理需确认其同意代表外国机构担任代表/联络人(Representative/liaison)。如果美国代理拒绝同意(或在 10 个工作日内未回复),则外国机构的官方通讯员/所有者/经营者(Official Correspondent/Owner Operator)将收到通知,并必须指定新的美国代理以履行监管义务。
美国代理的职责包括:
1,协助 FDA 与外国机构沟通(Foreign establishment);
2,回答有关外国机构/企业进口或拟进口到美国的医疗器械的问题;
3,协助 FDA 安排对外国机构的检查;
4,如果 FDA 无法直接或迅速联系到外国机构,FDA 可以向美国代理人提供信息或文件,此类行为应被视为等同于向外国机构提供相同的信息或文件。
美国代理必须居住在美国境内或在美国境内设有营业地点,不得使用邮政信箱(Post office box)或私信箱(P.O. box)作为地址,该地址必须是 FDA 工作人员可以访问其所在位置的实际地点。
FDA 不颁发任何类型的注册证书
参与生产和分销拟在美国使用的医疗器械的企业(Business)在 FDA 注册时,并不会收到 FDA 官方颁发的证书(Certificate)。



美国食品药品监督管理局 FDA 不颁发(Issue)任何类型的医疗器械机构(Device establishment)注册证书(Registration certificates),也不会为已注册机构并列名其医疗器械的设施的注册信息提供信息认证(Certify)。当某设施(Facility)在 FDA 注册并列名其医疗器械时,FDA 注册和列名数据库(Registration and listing database)中的相应条目(Entry)并不表示该设施(Facility)或其医疗器械已获得 FDA 的批准(Approval)、许可(Clearance)或授权(Authorization)。
据 FDA 介绍,一些公司在美国销售医疗器械时会附带“FDA 注册证书”(FDA registration certificates),这些证书通常看起来像官方政府文件,并可能带有 FDA 标识,然而,这并不属于真正意义上的 FDA 官方证书文件,FDA 并不颁发医疗器械注册证书(FDA does not issue device registration certificates)。
以上便是美国进口清关流程中关于 FDA 监管产品医疗器械机构注册和器械列名的流程介绍。












