




【美国进口清关】FDA 医疗器械:上市前通知 510(k) 和 510(k) 提交类型

 3285
3285

一键完成百千笔付款,超低费率+极速到账,一年轻松帮你省下数十万。
上市前通知(Premarket Notification)510(k) 适用于低至中等风险且存在合法上市对比器械(Legally marketed predicate device)的医疗器械,可向 FDA 提交的上市前通知 510(k) 有三种类型:传统 510(k)(Traditional)、特殊 510(k)(Special)和简化 510(k)(Abbreviated)。传统 510(k)(Traditional 510(k))适用于任何情况,依赖于实质等同 SE 的证明,而简化 510(k)(Abbreviated 510(k))和特殊 510(k)(Special 510(k))仅在满足特定条件时才可使用。由于 FDA 医疗器械与放射健康中心 CDRH(Center for Devices and Radiological Health)采取了各种措施,包括制定政策以明确和加强对 510(k) 申报内容(510(k) submission content)的要求,自 2009 年以来,510(k) 提交材料(510(k) submission)的平均页数增加了一倍多。
上市前通知(Premarket Notification)510(k)
介绍(Introduction)
凡打算在美国市场销售 I、II 和 III 类医疗器械(用于人类且无需提交上市前批准申请)的相关方,都必须向 FDA 提交 510(k) 申报,除非该医疗器械符合《联邦食品、药品和化妆品法案》(Federal Food, Drug, and Cosmetic Act)(FD&C 法案)规定的 510(k) 豁免要求,且未超出医疗器械分类法规章节 .9 中规定的豁免范围(例如,21 CFR 862.9、21 CFR 862.9)。目前,并没有统一的 510(K) 申请表格,但 21 CFR 807 子部分 E(21 CFR 807 Subpart E)明确了 510(k) 申请提交的要求。在医疗器械上市销售前,每位申报方都必须收到来自 FDA 的一封信函形式的命令,该命令认定该医疗器械实质等同 SE(Substantially equivalent),并声明该医疗器械可以在美国销售。此命令即为医疗器械可以进行商业分销(Commercial distribution)的“上市许可”(Clears)。
在申报方收到 FDA 声明医疗器械为实质等同 SE 的命令之前,该医疗器械不得上市销售。一旦该医疗器械被认定为实质等同 SE,即可在美国市场销售。SE 认定(SE determination)通常在 90 天内完成,并基于申报方提供的信息作出决定。
请注意,FDA 通常不进行 510(k) 预许可设施检查(510(k) pre-clearance facility inspections)。申报方可在获得 510(k) 许可(510(k) clearance)后立即将该医疗器械上市销售。制造商(Manufacturer)应在获得 510(k) 许可后随时准备接受 FDA 质量体系检查(Quality system inspection)(21 CFR 820)。
什么是实质等同 SE(Substantial Equivalence)
510(k) 申报要求证明该医疗器械与另一种在美国合法上市的医疗器械实质等同(Substantial equivalence)。
如果某医疗器械与“对比器械”相比,满足以下条件,则该医疗器械属于实质等同:
与“对比器械”(Predicate)具有相同的预期用途(Intended use);并且 具有与“对比器械”(Predicate)相同的技术特征(Technological characteristics);或 与“对比器械”(Predicate)具有相同的预期用途(Intended use);并且 具有不同的技术特征,且不会引起不同的安全性和有效性问题;并且 提交给 FDA 的信息表明该医疗器械与合法上市的医疗器械具有同等的安全性和有效性。
实质等同声明(A claim of substantial equivalence)并不意味着新型医疗器械与“对比器械”(Predicate)必须完全相同。FDA 首先确定新型医疗器械和“对比器械”(Predicate)具有相同的预期用途,且技术特性的任何差异均不会引起安全性和有效性方面的不同问题。然后,FDA 通过审查用于评估技术特性和性能数据差异的科学方法,确定该器械是否与“对比器械”(Predicate)具有同等的安全性和有效性。这些性能数据可能包括临床数据(Clinical data)和非临床台架性能数据(Non-clinical bench performance data),例如工程性能测试(Engineering performance testing)、无菌性(Sterility)、电磁兼容性(Electromagnetic compatibility)、软件验证(Software validation)、生物相容性评估(Biocompatibility evaluation)等。
申报方必须收到认定其医疗器械实质等同的信函,方可在美国上市销售。如果 FDA 认定某医疗器械不构成实质等同,申报方可以:
重新提交包含新数据的另一份 510(k) 申报, 通过 De Novo 分类流程申请 I 类或 II 类分类认定, 提交新分类申请,或 提交上市前批准申请 PMA。
谁需要提交 510(k) 申报(Who is Required to Submit a 510(k))
美国《联邦食品、药品和化妆品法案》(FD&C Act )和 510(k) 法规(510(k) regulation)(21 CFR 807)并未明确规定必须由谁提交 510(k) 申报,而是规定了哪些行为(例如将医疗器械引入美国市场)需要提交 510(k) 申报。
以下四类相关方(Categories of parties)必须向 FDA 提交 510(k) 申报:
1,美国国内制造商将医疗器械引入美国市场(Domestic manufacturers introducing a device to the U.S. market)
如果成品医疗器械制造商(Finished device manufacturers)按照其自身规格(Specifications)制造医疗器械并在美国销售,则必须提交 510(k) 申报(Submit a 510(k))。出售给最终用户的成品医疗器械配件也被视为成品医疗器械。但是,医疗器械组件制造商(Manufacturers of device components)无须提交 510(k) 申报,除非该组件作为替换零部件(Replacement parts)向最终用户进行推广销售。合同制造商(Contract manufacturers),即根据合同按照他人规格制造医疗器械的公司,无需提交 510(k) 申报。
2,产品规格制定方将医疗器械引入美国市场(Specification developers introducing a device to the U.S. market)
产品规格制定方负责制定成品医疗器械的规范,但该医疗器械的制造依据合同由其他公司或实体(Firm or entity)承担。规范开发人员提交 510(k),而非合同制造商(Contract manufacturer)。
3,对标签进行更改的重新包装商或重新贴标商(Repackers or relabelers),或其操作对医疗器械产生重大影响的重新包装商或重新贴标商
如果重新包装商或重新贴标商对标签进行重大更改或以其他方式影响医疗器械状况,则可能需要提交 510(k)。重大标签更改(Significant labeling changes)可能包括修改说明书(Modification of manuals),例如添加新的预期用途、删除或添加警告、禁忌症(Contraindications)等。灭菌等操作可能会改变医疗器械状况(Condition of the device)。但是,大多数重新包装商或重新贴标商无需提交 510(k)。
4,将医疗器械引入美国市场的外国制造商/出口商(Foreign manufacturers/exporters)或其美国代理(U.S. representatives)
请注意,所有 II 类和 III 类器械以及部分 I 类器械的制造商(包括产品规格制定方),在器械开发过程中均须遵守设计控制要求(Design controls)(21 CFR 820.30)。510(k) 持有方(The holder of a 510(k))在现场检查(Site inspection)期间必须提供设计控制文件以供 FDA 审查。此外,任何器械规格或制造工艺的变更(Device specifications or manufacturing processes)均须符合质量体系法规(Quality System regulation)(21 CFR 820)的规定,并可能需要提交新的 510(k)。
何时需要提交 510(k)(When a 510(k) is Required)
以下情况需要 510(k):
1,除非获得豁免,否则首次将医疗器械投入商业流通(Commercial distribution)(上市销售)时,必须遵守以下规定:自 1976 年 5 月 28 日(《医疗器械修正案》生效日)(Medical Device Amendments to the Act)起,任何希望在美国销售医疗器械的相关方,必须在产品上市前至少 90 天提交 510(k),即使该医疗器械在此日期前已处于研发或临床试验阶段(Under development or clinical investigation)。若你公司的医疗器械未在 1976 年 5 月 28 日前上市销售,则必须提交 510(k)。
2,合法上市的医疗器械发生了变更或修改(Change or modification),且该变更可能对其安全或有效性产生重大影响。510(k) 持有方有责任判断该修改是否会显著影响器械的安全性或有效性。任何修改(Modifications)都必须符合质量体系法规 21 CFR 820 的规定,并记录于器械主记录和变更控制记录中(Device master record and change control records)。FDA 建议将提交或不提交新 510(k) 的理由记录在变更控制记录中。
如果对现有医疗器械的变更或修改(Changes or modifications)可能会严重影响医疗器械的安全性或有效性,或者该医疗器械将用于新的或不同的预期用途,则需要提交新的 510(k)(A new 510(k) submission is required)。
何时不需要提交 510(k)(When a 510(k) is Not Required)
以下是不需要 510(k) 的示例:
1,你将未完成的医疗器械出售给其他公司(Firm)进行进一步加工,或出售组件(Components)其他公司组装医疗器械使用。但若你的组件将作为替换零部件(Replacement parts)直接销售给最终用户(End users),则需提交 510(k)。
2,你的医疗器械尚未上市或商业分销(Marketed or commercially distributed)。开发、评估或测试医疗器械(Develop, evaluate, or test a device)无需提交 510(k)。此规定同样适用于临床评估(Clinical evaluation)。请注意,如果你使用医疗器械开展临床试验(Clinical trials),则需遵守研究用医疗器械豁免 IDE(Investigational Device Exemption)法规(21 CFR 812)。
3,你分销(Distribute)另一家公司在美国国内制造的医疗器械。你可以在该医疗器械贴上“由 ABC 公司分销”(Distributed by ABC Firm)或“为 ABC 公司制造”(Manufactured for ABC Firm)的标签(21 CFR 801.1),并将其销售给最终用户,无需提交 510(k)。
4,在大多数情况下,如果你是重新包装商或重新贴标商(Repackers or relabelers),且医疗器械原有标签或状态未发生重大改变,则标签应与 510(k) 中提交的标签一致,并具有相同的适应症(Indications for use)、警告(Warnings)和禁忌症(Contraindications)。
5,你的医疗器械在 1976 年 5 月 28 日前已合法上市销售,且在设计、组件、制造方法或预期用途方面未发生重大变更或修改。此类医疗器械享有“祖父条款”(Grandfathered)权利,并且你拥有修订前状态(Preamendment Status)文件来证明这一点。
6,该医疗器械在美国境外制造,且你是该外国制造医疗器械的进口商。如果外国制造商(Foreign manufacturer)已提交 510(k) 并获得上市许可(Received marketing clearance),则无需再次提交 510(k)。外国制造商获得该医疗器械的 510(k) 许可(510(k) clearance)后,即可将其医疗器械出口给任何美国进口商。
7,根据法规 21 CFR 862-892,你的医疗器械可豁免 510(k)。也就是说,某些 I 类或 II 类器械首次上市时无需提交 510(k)。可以在“医疗器械豁免 510(k) 和 GMP 要求”(Medical Device Exemptions 510(k) and GMP Requirements)中找到 I 类和 II 类豁免器械的列表。但是,如果器械超出了器械分类法规章节(Device classification regulation chapters) .9 条款(如 21 CFR 862.9)规定的豁免范围,例如,该医疗器械的预期用途与同类已上市器械不同,或采用不同基础科学技术运作,或是经再处理的的一次性医疗器械(Single-use device),则必须提交 510(k) 才能上市。
修正案前医疗器械(Preamendment Devices)
修正案前医疗器械(Preamendments device)是指于 1976 年 5 月 28 日之前已在美国合法销售的医疗器械,且该医疗器械未曾:
自那时起,该产品已发生重大变化或修改(Significantly changed or modified);并且 FDA 尚未发布要求提交上市前批准申请(PMA application)的法规。
符合上述标准的医疗器械属于“祖父条款”(Grandfathered)医疗器械,无需提交 510(k)。该医疗器械的预期用途(Intended use)必须与 1976 年 5 月 28 日之前上市的医疗器械相同。若器械标签标注的预期用途发生变更,则该器械将被视为新型器械(New device),必须向 FDA 提交 510(k) 申报以获得上市许可(Marketing clearance)。
请注意,医疗器械必须在 1976 年 5 月 28 日前已合法上市销售才能适用“祖父条款”(Grandfathered)权利。如果你的医疗器械与适用“祖父条款”(Grandfathered)权利的医疗器械类似,且在 1976 年 5 月 28 日之后上市,则不符合“祖父条款”(Grandfathered)权利的要求,必须提交 510(k)。企业(Firm)若主张其医疗器械属于修正案前医疗器械(Preamendment Devices),则必须证明其器械已针对特定预期用途进行标记、推广和在州际贸易中分销,且该预期用途未发生改变。
第三方审查计划(510(k) Third Party Review Program)
FDA 医疗器械与放射健康中心 CDRH(Center for Devices and Radiological Health)已实施第三方审查计划(Third Party Review Program)。该计划为某些医疗器械制造商提供了一个选择,使其可将 510(k) 申报提交给 FDA 指定的私营机构(认可第三方)(Recognized Third Parties)进行审查,而非直接提交至 CDRH。根据该计划,第三方审查机构(Third Party Review Organizations)对符合条件的低风险医疗器械的 510(k) 申报(510(k) submissions)进行等同于 FDA 的审查(FDA-equivalent reviews)。
第三方审查计划是一项自愿的替代性审查流程(Voluntary alternative review process)。它允许经过认可(Accredited)的第三方审查机构代替 FDA 对 510(k) 申报进行审查。FDA 可以集中更多资源用于高风险和复杂医疗器械的审查,同时仍然确保第三方审查的低风险、低复杂医疗器械安全有效。最终,新的医疗产品可以更快地进入市场并惠及美国患者。
第三方审查计划包含两个关键部分:首先是第三方机构的认证(Accreditation of Third Parties),其次是 510(k) 审查本身(510(k) reviews)。该计划的第一部分,即认证(Accreditation),是指 FDA 如何确保第三方审查机构能够对符合条件的医疗器械进行与 FDA 同等水平的审查。一旦获得 FDA 认证,第三方审查机构即可自行执行该计划的第二部分:510(k) 审查。在第三方审查计划中,510(k) 申报方可以选择将其 510(k) 申报材料提交至第三方审查机构而非 FDA。第三方机构会将完成的审查报告及建议连同原始 510(k) 申报材料(Original 510(k) submission)一并转交 FDA。
根据相关法律规定,FDA 必须在收到第三方建议后 30 天内作出最终决定(Final decision)。
对比器械(Predicate device)
对比器械(Predicate device)是已通过 510(k) 流程(510(k) process)获准上市的合法医疗器械(Legally marketed device),用于将新型医疗器械与之进行比较以确定实质等同(Substantial equivalence)(21 CFR 807.92(a)(3))。虽然制造商可以指定多个对比器械,但 FDA 建议制造商确定一个对比器械,以简化和促进决策过程。
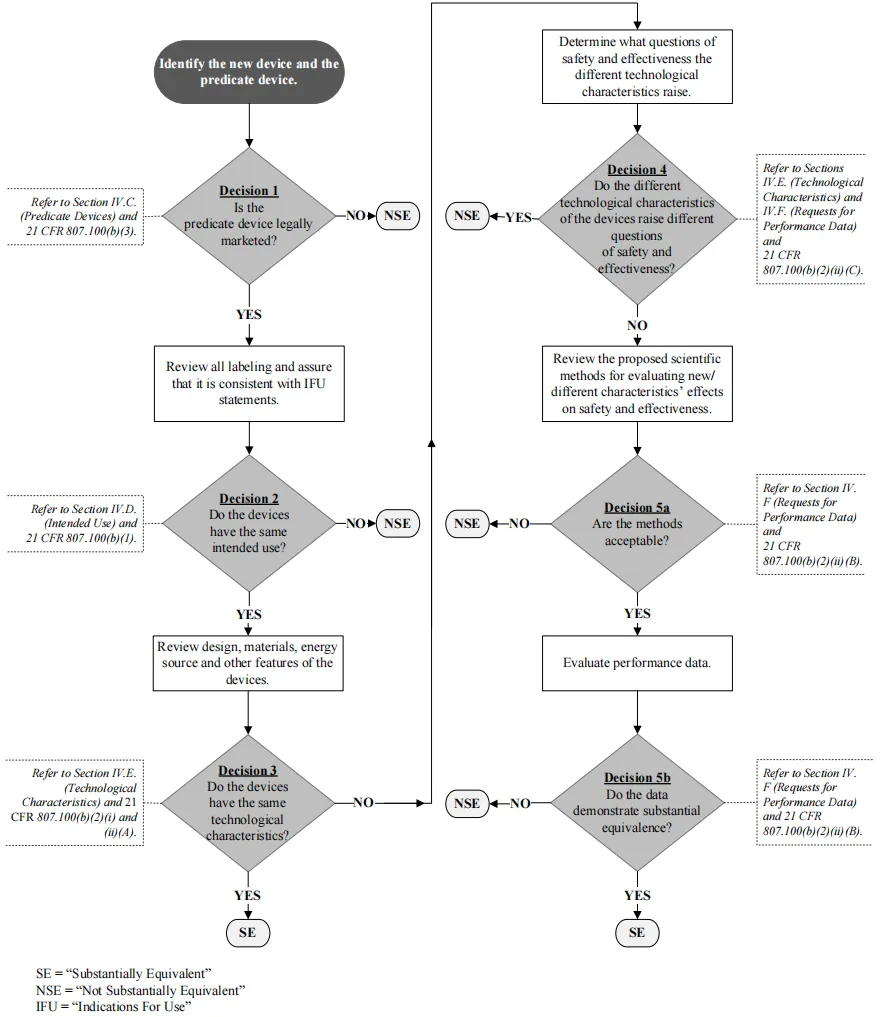
《联邦食品、药品和化妆品法案》第 513(i) 条(Section 513(i) of the FD&C Act)和 21 CFR 807.100(b) 规定,新型器械要被视为与对比器械实质等同 SE(Substantially equivalent),则必须满足以下条件:新型器械必须与对比器械具有相同的预期用途(Intended use)和相同的技术特性(Technological characteristics),或者具有不同的技术特性,但这些特性不会引发与对比器械不同的安全性和有效性问题。
介绍(Introduction)
上市前通知(Premarket Notification)510(k) 是向 FDA 提交的上市前申报(Premarketing submission),用于证明拟上市医疗器械与未受上市前批准 PMA 约束的已合法上市(Legally marketed)医疗器械(即“对比器械”)(Predicate device)实质等同 SE(Substantial equivalence),从而证明该医疗器械的安全性和有效性。申报方必须将其 510(k) 申报医疗器械与在美国境内合法上市的类似医疗器械进行比对。近期通过 510(k) 流程获得许可(Cleared)的医疗器械通常用作对比器械。但是,任何在美国合法上市的医疗器械均可用作对比器械。这包括:通过 510(k) 流程获得许可(Cleared)的医疗器械;1976 年 5 月 28 日之前已合法上市(Legally marketed)的医疗器械(修正案前医疗器械)(Preamendments device);最初作为 III 类医疗器械在美国市场上销售(上市前批准)(Premarket Approval)后降级为 II 类或 I 类的医疗器械;或 510(k) 豁免医疗器械(510(k) exempt device)。
实质等同声明(A claim of substantial equivalence)并不要求医疗器械必须完全相同。实质等同 SE 基于以下方面确立:预期用途(Intended use)、设计(Design)、使用或输送的能量(Energy used or delivered)、材料(Materials)、性能(Performance)、安全性(Safety)、有效性(Effectiveness)、标签标识(Labeling)、生物相容性(Biocompatibility)、标准(Standards)和其他适用特性(Applicable characteristics)。你可以对修正案前或修正案后合法上市的医疗器械主张实质等同。合法上市(Legally marketed)意味着对比器械(Predicate)不能违反或曾经违反《联邦食品、药品和化妆品法案》(FD&C Act)。申报方可以对已不再在美国销售的医疗器械主张实质等同。
修正案后医疗器械(Postamendments Device)
修订后器械是指 1976 年 5 月 28 日之后上市的医疗器械。据 FDA 介绍,由于医疗技术自 1976 年以来发生了巨大变化,几乎所有 510(k) 申报(510(k) submissions)都主张(Claim)与最近根据 510(k) 流程获得许可(Cleared)的修正案后器械具有实质等同性。
修正案前医疗器械(Preamendments Device)
修正案前器械是指于 1976 年 5 月 28 日之前已在美国合法上市(Legally marketed)的医疗器械,且未:
自那时起发送重大变更或修改(Significantly changed or modified);并且 FDA 尚未发布要求上市前批准 PMA 提交的法规。
符合上述标准的医疗器械被称为“祖父条款”(Grandfathered)器械,无需提交 510(k)。该医疗器械的预期用途必须与 1976 年 5 月 28 日之前上市的医疗器械相同。若该医疗器械的标签上注明了新的预期用途,则该器械将被视为新型医疗器械(New device),必须向 FDA 提交 510(k) 以获得上市许可(Marketing clearance)。
企业(Firm)若要主张(Claim)其医疗器械为修正案前器械(Preamendments device),则必须证明该医疗器械在州际贸易中被标注(Labeled)、推广(Promoted)和分销(Distributed)时具有特定预期用途(Specific intended use),且该预期用途未发生改变。
如果使用修正案前器械作为对比器械(Predicate device),可能需要提供其符合修正案前状态标准的证明文件。修正案前器械不会获得 510(k) 编号(510(k) number),因为其不受 510(k) 要求的约束。
如何搜索对比器械(How to Search for a Predicate Device)
FDA 510(k) 数据库包含所有根据 510(k) 流程获得许可(Cleared)的医疗器械。FDA 官网数据库通常于每月 5 日左右更新。医疗器械分类和产品代码对于搜索对比器械至关重要。可以通过在产品代码分类数据库(Product Code Classification Database)中进行搜索来查找你的医疗器械分类信息。如果你的器械类型(Device type)已获得 FDA 的最终分类(Final classification)(例如,21 CFR 888.1100,关节镜),分类数据库将提供医疗器械分类专业组(Classification panel)(例如,骨科器械)、通用名称(Common name)、产品代码(Product code)和 CFR 法规(CFR regulation)信息。
有助于查找对比器械(Predicate device)的信息包括:
类似器械(Similar devices)的名称 - 器械(Marketed)上市销售时所用的商业名称(Traded name); 类似器械的制造商; 上市状态(Marketing status),即修正案前或修正案后器械; 修正案后器械的 510(k) 编号; 分类信息(Classification information),即产品代码、分类法规等。
FDA 会为每个通用类别(Generic category)的医疗器械分配一个唯一的三字母产品代码(Product code),无论该器械是否已被 FDA 正式分类。只有 FDA 生成的产品代码才能与 510(k) 和其他 FDA 数据库兼容。虽然并非绝对,但类似器械类型(Similar device types)的 510(k) 通常在 510(k) 数据库中通过相同的产品代码进行关联。因此,通过产品代码搜索参照器械通常是最有效的(其他检索框可留空)。
或者,如果你知道类似器械(Similar device)的制造商名称,可以按制造商名称搜索数据库。请注意,510(k) 数据库仅包含原始申请信息(Original application information)。也就是说,510(k) 数据库保留原始申请人(Original applicant)的名称以及 510(k) 中提供的原始商品名称(Original trade name)。它不会更新以反映产品的当前所有者或分销商信息,也不会更新商品名称的任何变更。
510(k) 上市前通知数据库(510(k) Premarket Notification):https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/pmn.cfm
如何有效使用对比器械(How to Effectively Use Predicate Devices)
制造商必须确定一个主要对比器械(Primary predicate device),该器械在适应症(Indications for use)和技术特性(Technological characteristics)方面与 510(k) 申报中的待审器械(Under review)最为相似。在某些情况下(Certain circumstances),制造商可确定多个对比器械,以协助证明实质等同 SE(Substantial equivalence)。使用分拆对比器械的做法不符合 510(k) 监管标准(510(k) regulatory standard)。制造商可以在其 510(k) 中确定“参考器械”(Reference devices),以支持科学方法或标准参考值(Scientific methodology or standard reference values)。
510(k) 申报流程(510(k) Submission Process)
确认收讫(Acknowledgement of Receipt)
可以通过发送 eSTAR 或 eCopy 来提交你的 510(k)。
美国食品药品监督管理局 FDA 收到你提交的 510(k) 后,会为其分配一个唯一的控制编号(Control number),该编号通常被称为 510(k) 编号(510(k) number)或 K 编号(K number)。510(k) 编号以字母 K 开头,后跟 6 位数字,前 2 位数字表示收到 510(k) 的年份,后四位数字则表示该年度的提交编号(Submission number),从 0001 开始递增。
例如,2025 年提交的第一份 510(k) 的编号是 K250001。
然后,FDA 会进行两项验证检查,以确认:
1,已收到该份提交的 510(k) 的用户费用(User fee)的付款。用户费用金额以 FDA 收到 510(k) 的日期为准,而非申报方发送 510(k) 的日期。
2,已提供有效的 510(k) 的 eSTAR 或 eCopy。
如果未支付相应的用户费用(Proper user fee)和/或未提供有效的 eSTAR 或 eCopy,FDA 通常会在收到 510(k) 后 7 天内,通过电子邮件向 510(k) 提交人发送一封“暂停函”(Hold Letter)。申报方须自“暂停函”通知发出之日起 180 个日历日内,解决用户费用或 510(k) 提交(Submission)方面的问题。如果问题在 180 天内未得到解决,则提交的 510(k) 将被视为撤销(Withdrawn)并从 FDA 的审核系统(Review system)中删除,申报方需要提交一份全新且完整的 510(k) 以获得 FDA 的上市许可(Marketing clearance)。
如果已支付相应的用户费用并提供了有效的 eSTAR 或 eCopy,FDA 将通过电子邮件向提交的 510(k) 中指定的联系人发送确认函(Acknowledgement Letter)。该确认函中将注明:
收到日期(即 FDA 收到提交的 510(k)、相应的用户费用支付以及有效的 eSTAR 或 eCopy 的日期);以及 FDA 分配的 510(k) 编号(510(k) number)
请注意:确认函(Acknowledgement Letter)并非 FDA 上市许可函(Marketing clearance letter)。在后续与 FDA 就 510(k) 事宜进行的所有通信中,均应注明确认函中列出的 510(k) 编号。未注明 510(k) 编号可能会导致处理延误。
在上市前提交(Premarket submission)的审查过程中,美国食品药品监督管理局 FDA 惯常通过两种方式与申请人沟通:一是正式函件(如重大缺陷通知函(Major Deficiency Letter)或通过信函、电话、传真或电子邮件发出的补充信息请求,并以后续函件确认暂缓状态);二是采用交互式审查流程。
格式和申报材料的受理审查(Format and Submission Acceptance Review)
对于 eSTAR,鉴于使用电子提交模板(Electronic submission template)正确准备的电子提交应代表完整的提交(Complete submission),因此 eSTAR 提交预计不会经历拒绝接受 RTA(Refuse to accept)流程。然而,FDA 计划对 eSTAR 实施病毒扫描和技术筛查流程(Virus scanning and technical screening process)。
如果 eSTAR 提交(eSTAR submission)不完整,FDA 将通过电子邮件通知申报方并指明缺失信息,该 510(k) 将被置于待处理状态(Remain on hold)直至提交完整的替换 eSTAR(Replacement eSTAR)。如果在技术审查缺陷通知(Technical screening deficiency notification)之日起 180 天内未收到替换 eSTAR,FDA 将视该 510(k) 已撤销(Withdrawn),并在系统中关闭该提交记录。
对于 eCopy,FDA 审查专员(Lead Reviewer)将根据《510(k) 拒绝受理政策指南》(Refuse to Accept Policy for 510(k)s)中相应的受理性审核清单(Acceptance Checklist)进行受理性审查(Acceptance Review)。
确认函发出后,FDA 将根据提交的 510(k) 中登记的医疗器械类型和医疗专业领域(Device type and medical specialty) ,将该 510(k) 转交至相应的医疗器械和放射健康中心 CDRH 的产品评估和质量办公室 OPEQ 的下属办公室 OHT。
办公室收到 510(k) 后,会将其转交至相应的部门(Appropriate Division),随后分配给审查专员(Lead Reviewer)处理。
在受理性审查(Acceptance Review)过程中,审查专员(Lead Reviewer)将确定提交的 510(k) 是否满足最低可受理性标准,并决定是否应接受实质性审查(Substantive review)。
在收到 510(k) 申报的 15 天内,申报方将收到关于受理性审查(Acceptance Review)结果的电子通知,该通知将:
注明被分配至该 510(k) 的 FDA 审查专员的姓名和联系信息 并说明该 510(k) 申报的状态
受理性审查结果将为以下之一:
510(k) 已接受实质性审查;或 510(k) 未获受理审查(即被视为拒绝受理或 RTA);或 由于 FDA 未在 15 个日历日内完成受理性审查,510(k) 正在接受实质性审查。
未获受理审查的 510(k) 将被置于 RTA 暂缓处理状态(RTA Hold),申报方需在 180 个日历日内全面解决 RTA 暂缓通知中列出的缺陷。若未能完成,则该提交的 510(k) 将被视为撤销(Withdrawn)并从 FDA 的审核系统(Review system)中删除,申报方需要提交一份全新且完整的 510(k) 以获得该医疗器械 FDA 的上市许可(Marketing clearance)。
获受理后(Once accepted),510(k) 将进入实质性审查阶段(Substantive Review)。
实质性审查(包括实质性互动和互动式审查)(Substantive Review (including Substantive Interaction and Interactive Review))
在实质性审查期间,审查专员(Lead Reviewer)对提交的 510(k)(510(k) submission)进行全面审查(Comprehensive review),并通过实质性互动(Substantive Interaction)与申报方进行沟通,这应在收到 510(k) 申报后的 60 个日历日内进行。
实质性互动沟通通常包括:
一封电子邮件,声明 FDA 将通过互动式审查(Interactive Review)解决所有未决缺陷;或 补充信息请求(Additional Information request),将该 510(k) 置于暂缓处理状态(Hold)
互动式审查(Interactive Review)
如果审查专员选择继续进行互动式审查,则表示审查专员已确定任何未决缺陷均可在医疗器械用户费用修正案 MDUFA(Medical Device User Fee Amendment)规定的期限内得到充分解决,且该 510(k) 不会被置于暂缓处理状态(Placed on hold)。在互动式审查期间,审查专员将通过一下工具与申报方进行沟通:
电子邮件(Email) 电话(Telephone Call)
在互动式审查期间,审查专员可能要求申报方提供补充信息,申报方可将信息直接发送给审查专员,也可以发送给 FDA。请注意:在互动式审查期间,提交给 FDA 的任何信息都必须包含有效的 eSTAR 或 eCopy。
补充信息请求(Additional Information (AI) Request)
FDA 指定医疗器械上市前申请决策(Medical device marketing application)所需的补充信息请求(FDA request for additional information)被称为“缺陷通知”(Deficiency)。
如果审查专员发出 AI 请求,该 510(k) 将被暂缓处理(Placed on hold)。申报方需在 AI 请求发出之日起 180 个日历日内提交完整的 AI 请求回复。请注意:FDA 必须在 AI 请求之日起 180 个日历日内收到完整的回复。超过 180 天的延期不予批准。若 FDA 在 AI 请求发出之日起 180 天内未收到针对 AI 请求中所有未决缺陷的完整回复,则该提交的 510(k) 将被视为撤销(Withdrawn)并从 FDA 的审核系统(Review system)中删除,申报方需要提交一份全新且完整的 510(k) 以获得该医疗器械 FDA 的上市许可(Marketing clearance)。
申办方必须向 FDA 提交回复,并附上有效的 eSTAR 或 eCopy。回复内容应包括:
提交人姓名(Submitter's name); 510(k) 编号(510(k) number); 提交内容作为 510(k) 补充信息(AI)的标识(Identification); FDA 要求提供补充信息的日期;以及 按条理清晰的方式呈现所要求的补充信息。
510(k) 决定函(510(k) Decision Letter)
FDA 对 510(k) 作出 MDUFA 决定的目标时限为 90 个 FDA 工作日。FDA 工作日的计算方式为:从 510(k) 收到日期到 MDUFA 决定日期之间的日历天数,且不包含因要求补充信息(AI)而暂缓处理的天数。MDUFA 对 510(k) 提交的决定包括实质等同 SE(Substantially equivalent)或非实质等同 NSE(Not substantially equivalent)的认定结果。
当做出决定时,FDA 将通过电子邮件向申报方发出决定函(Decision letter),电子邮件地址为 510(k) 封面函(510(k) cover letter)中提供的邮件地址(Email address)。
获得 SE 决定的 510(k) 被视为“已获上市许可”(Cleared)。FDA 每周都会将已获许可的 510(k)(Cleared 510(k))添加到 510(k) 数据库中。适应症 IFU(Indications for Use)和 510(k) 摘要(510(k) Summary)或 510(k) 声明(510(k) Statement)将作为 SE 函件(SE letter)的附件包含在内。IFU 和 510(k) 摘要或声明构成 SE 文件包(SE package)。完整的 SE 文件包每月发布一次。由于 IFU 被视为 SE 函件的附件,故无需单独签名。因此,SE 函件上的签名将同时适用于该函件和 IFU。
如果 FDA 未能在 100 个 FDA 工作日内(即 MDUFA 目标日期后 10 天)做出 MDUFA 决定,则将发布《未达 MDUFA 期限通知》(Missed MDUFA Communication)。这是一份发送给申报方的书面反馈(Written feedback),供后续会议或电话会议讨论,内容包括主要未决审查事项或阻碍 FDA 做出最终决定的其他原因,以及预计完成日期。
与 510(k) 申报方沟通的时间表(Timeline of Communication with 510(k) Submitters)
FDA 遵循 MDUFA 绩效目标(MDUFA performance goals)来审查提交的 510(k)(510(k) submissions)。
当你向医疗器械与放射健康中心 CDRH 提交 510(k)(510(k) submission)进行审查时,你的官方通讯员(Official correspondent)可通过 FDA 在线申报系统实时追踪 FDA 的审查进度。
510(k) 申报类型(510(k) Submission Programs)
概述(Overview)
申报方可以从三种上市前通知(Premarket Notification)510(k) 提交类型中选择,以便在适合根据 510(k) 计划进行审查时为其医疗器械寻求上市许可(Marketing clearance):传统(Traditional)、特殊(Special)和简化(Abbreviated)。
传统 510(k) 申报计划(Traditional Program)是 21 CFR 807 规定的原始申报类型(Original submission type)。1998 年,FDA 制定了特殊 510(k) 计划(Special 510(k) Programs)和简化 510(k) 计划(Abbreviated 510(k) Programs),以方便审查某些符合 510(k) 要求的申报类型。当 510(k) 申报符合某些条件时,可以使用特殊 510(k) 和简化 510(k) 申报类型。传统、特殊和简化 510(k) 申报计划的用户费用相同。
传统 510(k)(Traditional 510(k))
传统 510(k) 可用于任何原始 510(k),或用于对先前依据 510(k) 获得许可的医疗器械所作的变更,FDA 通常会在收到传统 510(k) 申报(Traditional 510(k) submissions)后 90 天内进行审查。
特殊 510(k)(Special 510(k))
如果评估变更的方法已完善,且变更结果能够以摘要或风险分析的形式进行充分审查,则医疗器械制造商可以选择提交特殊 510(k) 来处理其现有医疗器械的变更。
正如 FDA《特殊 510(k) 计划》(The Special 510(k) Program)指南中所述,在以下情况下,对现有医疗器械进行设计或标签变更(包括对适应症(Indications for use)的某些变更)可能适用于特殊 510(k):
1,拟议变更由获得合法授权销售现有医疗器械的制造商提交;
2,无需提供绩效数据,或者如果需要,则有成熟的方法来评估变更;
3,所有支持实质等同 SE 所需的绩效数据均可采用摘要或风险分析(Summary or risk analysis)的形式进行审查。
FDA 通常会在收到特殊 510(k) 申报(Special 510(k) submissions)后 30 天内进行审查。
简化 510(k)(Abbreviated 510(k))
当 510(k) 申报依赖于以下条件时,医疗器械制造商可以选择提交简化 510(k):
FDA 指南文件(FDA guidance document(s)) 符合该医疗器械类型特殊管制(Special control(s))要求的证明,或 自愿性共识标准(Voluntary consensus standard(s))
简化 510(k) 申报(Abbreviated 510(k) submission)必须包含 21 CFR 807.87 传统 510(k) 中规定的必要要素(Required elements)。然而,在简化 510(k) 申报中,企业可选择提供关于指南文件和/或特殊管制要求(Special controls)或符合 FDA 公认标准的声明使用情况的摘要报告,以方便 FDA 审核。如果制造商提交的简化 510(k) 申报依赖于自愿性共识标准的普遍使用(General use of a voluntary consensus standard),则应包含此类使用的依据以及支持该标准使用方式的基础信息或数据。
FDA 通常会在收到简化 510(k) 申报(Abbreviated 510(k) submissions)后 90 天内进行审查。
如何准备特殊 510(k)(How To Prepare A Special 510(k))
概述(Overview)
可向 FDA 提交三种类型的上市前通知(Premarket Notification)510(k)。
特殊 510(k) 计划(The Special 510(k) Program)旨在促进制造商对其自有合法上市(Legally marketed)的对比器械(Predicate device)“现有器械”(Existing device)所作变更的提交、审查和许可(Clearance)。此类医疗器械已通过 510(k) 许可(510(k) clearance)、修正案前状态(Preamendments status)、重新分类(Reclassification)、或根据《联邦食品、药品和化妆品法案》513(f)(2) 条(Section 513(f)(2) of the FD&C Act)获得批准(Granted)的 De Novo 分类申请(Classification request)获得商业分销(Commercial distribution)授权。
对于某些医疗器械变更,FDA 认为设计控制程序(Design control procedures)可以产生可靠的结果,这些结果可作为实质等同 SE(Substantial equivalence)判定的基础,同时不会影响实质等同的法定和监管标准。根据设计控制(Design controls)的规定,制造商必须执行验证和确认(Conduct verification and validation)流程(21 CFR 820.30(f) 和 (g))。验证和确认包括确保设计输出符合设计输入,以及医疗器械符合既定用户需求和预期用途的程序。质量体系法规 QS(21 CFR Part 820)规定了适用于受设计控制(21 CFR 820.30 和 820.180)约束的设计变更的记录建立和维护要求。根据《联邦食品、药品和化妆品法案》第 704(e) 条(Section 704(e) of the FD&C Act)的规定,此类记录必须应 FDA 调查员(FDA investigator)的要求提供。
背景(Background)
美国食品药品监督管理局 FDA 于 1998 年设立了特殊 510(k) 计划(Special 510(k) Program),并在指南文件《新 510(k) 范式:上市前通知中证明实质等同的替代方法》(The New 510(k) Paradigm: Alternate Approaches to Demonstrating Substantial Equivalence in Premarket Notifications)(简称“新 510(k) 范式指南”)(New 510(k) Paradigm Guidance)中描述了该计划及其相关政策。该计划旨在为符合 510(k) 申报要求的某些变更创建高效的审查流程(Review process)。
设计控制(Design controls)要求已纳入质量体系法规(QS Regulation),并自 1997 年 6 月 1 日起生效(21 CFR 820.30、61 FR 52602)。特殊 510(k) 计划(Special 510(k) Program)利用设计控制要求,通过依赖风险分析以及对现有医疗器械的验证和确认来支持 SE 判定。特殊 510(k) 允许 FDA 和行业(Industry)在适当情况下依赖机构(Agency)先前对详细信息的审查,而无需更改与《联邦食品、药品和化妆品法案》第 510 和 513 条(Sections 510 and 513 of the FD&C Act)以及 21 CFR 807 子部分 E 下(21 CFR 807 Subpart E)的上市前通知流程相关的任何法定或监管要求。特殊 510(k) 计划提供了一种最少负担的申报途径(Least burdensome approach)用于审查制造商自身合法上市(Legally marketed)的对比器械(Predicate device)(“现有器械”)(Existing device)的某些变更,因为特殊 510(k) 计划为制造商提供了一条高效途径(Efficient pathway),使其能够提供为修改后的器械确立 SE 所需的最低限度信息(Minimum required information)。鉴于此效率优势,FDA 计划在文件控制中心 DCC(Document Control Center)特殊 510(k) 提交后 30 天内处理,而不是《联邦食品、药品和化妆品法案》第 510(n)(1) 条(Section 510(n)(1) of the FD&C Act)规定的 90 天。
FDA 目前针对特殊 510(k) 计划的监管策略,重点关注以下两点:用于评估变更的方法是否完善可靠,以及相关结果能否通过摘要或风险分析形式(Summary or risk analysis format)进行充分审查。
特殊 510(k) 仍需遵守 510(k) 申报(510(k) submissions)、510(k) 摘要(510(k) summary)或 510(k) 声明(510(k) statement)以及 III 类认证的内容和格式要求(分别为 21 CFR 807.87、807.90、807.92、807.93 和 807.94)。
何时选择特殊 510(k)(When to Choose a Special 510(k))
如果变更(Change)需要提交新的 510(k),则设计控制流程(Design control process)产生的摘要信息(Summary information)可与 21 CFR 807.87 中规定的 510(k) 必备要素(Required elements)共同作为实质等同 SE(Substantial equivalence)的依据(Basis)。
在以下情况下,对现有医疗器械进行设计或标签变更(包括对适应症(Indications for use)的某些变更)可能适用于特殊 510(k):
1,拟议变更由获得合法授权销售现有医疗器械的制造商提交;
2,无需提供性能数据(Performance data),或者如果需要,则有成熟的方法来评估变更;
3,所有支持实质等同 SE 所需的性能数据均可采用摘要或风险分析(Summary or risk analysis)的形式进行审查。
如果 FDA 认定特殊 510(k) 不适用于已提交的审查,FDA 将通知申报方该决定,并将该 510(k) 转换为传统 510(k)(Traditional 510(k))。转换后的 510(k) 仍以原受理日期作为审查期的开始日期。
如何提交 510(k)(How to Submit a 510(k))
自 2023 年 10 月 1 日起,所有 510(k) 申报(除非获得豁免)都必须使用 eSTAR 以电子方式提交。
特殊 510(k) 用户费用(User Fees for a Special 510(k))
传统(Traditional)、特殊(Special)和简化(Abbreviated)510(k) 的用户费用(510(k) user fee)相同。
如何准备传统 510(k)(How to Prepare a Traditional 510(k))
概述(Overview)
可以向 FDA 提交三种类型的上市前通知(Premarket Notification)510(k):传统 510(k)(Traditional),特殊 510(k)(Special)和简化 510(k)(Abbreviated)。1998 年,FDA 制定了特殊型和简化型 510(k) 计划,旨在简化特定类型 510(k) 申报的审查流程。当 510(k) 申报符合某些条件时,可以使用特殊 510(k) 和简化 510(k) 提交类型。
传统 510(k) 计划(Traditional 510(k) Program)适用于任何情况,以通过 510(k) 计划(510(k) Program)寻求医疗器械的上市许可(Marketing authorization)。
没有官方的 FDA 上市前通知(Premarket Notification)510(k) 表格。510(k) 是一份包含 21 CFR 807.87 法规所规定必须信息的提交文件。所有 510(k) 提交均基于与合法上市器械(亦称对比器械)的实质等同 SE(Substantial equivalence)原则。每份 510(k) 提交都需提供拟上市器械与对比器械之间的对比分析。
查找对比器械(Find a Predicate Device)
你应该确定一个与你计划通过 510(k) 计划提交的医疗器械在适用范围和技术特性方面最相似的主要对比器械(Primary predicate device)。在某些情况下,你可对多个对比器械主张实质等同 SE。
如何提交 510(k)(How to Submit a 510(k))
自 2023 年 10 月 1 日起,所有 510(k) 申报(除非获得豁免)都必须使用 eSTAR 以电子方式提交。
如何准备简化 510(k)(How to Prepare an Abbreviated 510(k))
何时选择简化 510(k)(When to choose an Abbreviated 510(k))
简化 510(k) 依赖于指南文件(Use of guidance documents),特殊管制要求(Special controls)和自愿共识标准(Voluntary consensus standards)。
简化 510(k) 申报必须包含 21 CFR 807.87(上市前通知提交所需信息)中规定的必要要素。在某些情况下(Under certain conditions),申报方在提交简化510(k) 时可能无需提交测试数据(Test data)。
当 510(k) 申报依赖于以下条件时,医疗器械制造商(Device manufacturers)可以选择提交简化 510(k)(Abbreviated 510(k)):
FDA 指南文件; 证明符合该医疗器械类型的特殊管制要求;或 自愿共识标准。
在简化 510(k) 提交中,制造商需依据指南文件和/或特殊管制要求的应用情况或声明符合公认共识标准提交摘要报告(Summary reports),以协助 FDA 对 510(k) 提交进行审查。
若 FDA 认定提交的简化 510(k) 不适用于 510(k) 审核(Review),FDA 将通知此决定,并将该 510(k) 转换为传统 510(k)(Traditional 510(k))。转换后的 510(k) 文件仍以原始收据日期作为审核期的开始日期。制造商应注意,在大多数情况下,转换后的 510(k) 申报需要提供更多补充信息。
特殊管制要求(Special Controls)
特殊管制要求(Special controls)是合理保证 II 类医疗器械(Class II device)安全性和有效性的一种方式。根据《联邦食品、药品和化妆品法案》第513(a)(1)(B) 条(Section 513(a)(1)(B) of the Federal Food, Drug, & Cosmetic Act),特殊管制要求(Special controls)的定义为:通过实施性能标准、上市后监督、患者登记、制定并发布指南(包括临床数据提交指南)、提出建议及其他适当措施,为医疗器械的安全性和有效性提供合理保障。医疗器械的分类法规(Classification regulation)中列明了该器械的特殊管制要求(如有)。医疗器械分类法规载于《联邦法规汇编》第 21 编第 862 至 892 部分(CFR part 862 through 892),也可以通过可搜索的产品分类数据库查询。
依赖特殊管制要求的简化 510(k) 应包含一份摘要报告(Summary report),说明对特殊管制要求的遵循情况以及医疗器械如何符合这些特殊管制要求,包括如何应用特殊管制要求来应对特定风险或问题。摘要报告应包含制造商为遵守特殊管制要求所做努力的相关信息,并概述任何偏差。
自愿共识标准(Voluntary Consensus Standards)
除了指南文件和特殊管制要求外,FDA 医疗器械与放射健康中心 CDRH 还致力于认可个别共识标准。
自愿共识标准(Voluntary consensus standard)是由美国国内和国际标准开发组织 SDOs(Standards Development Organizations)根据严格的共识原则制定或采用的标准,为活动或其结果提供可普遍重复使用的规则、指南或特征,旨在在特定情况下实现最佳秩序。
FDA 有权认可全部或部分国家和国际标准。认可的标准可以在指南文件或法规文件(例如描述与某种器械相关的特定风险的规则或命令)的序言中引用。
提交基于自愿共识标准的简化 510(k) 的医疗器械制造商,应提供《简化 510(k) 计划》附录 A(Appendix A of The Abbreviated 510(k) Program)中所述的信息,并按照指南文件《医疗器械上市前提交中自愿共识标准的适当使用》(Appropriate Use of Voluntary Consensus Standards in Premarket Submissions for Medical Devices)的建议提供符合性声明(Declaration of conformity)。
制造商提交基于自愿共识标准的一般应用(General use)的简化 510(k) 时,应说明该应用依据,并附上支持该标准应用方式的基础信息或数据。
如何提交 510(k)(How to Submit a 510(k))
自 2023 年 10 月 1 日起,所有 510(k) 申报(除非获得豁免)都必须使用 eSTAR 以电子方式提交。
基于安全和性能的路径(Safety and Performance Based Pathway)
概述(Overview)
可以向 FDA 提交 3 种类型的上市前通知(Premarket Notification)510(k):传统型(Traditional)、特殊型(Special)和简化型(Abbreviated)。基于安全和性能的路径是对简化 510(k) 路径(Abbreviated 510(k) pathway)的扩展,适用于某些已广为人知的医疗器械类型。
根据基于安全和性能的路径,医疗器械制造商可以选择采用 FDA 认同(FDA-identified)的性能标准(Performance criteria)来证明其医疗器械与对比器械具有同等的安全性和有效性。采用此路径不影响 FDA 依据法规或条例获取信息的权限。
哪些类型的医疗器械适合基于安全和性能的路径(What device types are appropriate for the Safety and Performance Based Pathway)
当 FDA 确定以下情况时,基于安全和性能的路径是适用的:
新型器械具有与已确定的对比器械相同的适应症(Indications for use),且其技术特性不会引发与已确定的对比器械不同的安全性和有效性问题;并且 新型器械符合 FDA 确定的所有性能标准(Performance criteria)。
如果上述任何条件未满足,申报方可以选择提交传统、特殊或简化的 510(k)。
常规 Foley 导尿管(Conventional Foley Catheters) 用于记录目的的皮肤电极(Cutaneous Electrodes for Recording Purposes) 牙科气动手持件及气动马达(Dental Air-Powered Dental Handpieces and Air Motors) 牙科粘结剂(Dental Cements) 牙科陶瓷(Dental Ceramics) 牙科印模材料(Dental Impression Materials) 义齿基托树脂(Denture Base Resin) 骨内种植体与骨内种植体基台(Endosseous Dental Implants and Endosseous Dental Implant Abutments) 小平面螺钉系统(Facet Screw Systems) 骨折固定板(Fracture Fixation Plates) 核磁共振接收专用线圈(Magnetic Resonance Receive-Only Coils) 骨科非脊柱金属接骨螺钉及垫圈(Orthopedic Non-Spinal Metallic Bone Screws and Washers) 软性(亲水性)日常佩戴型隐形眼镜(Soft (Hydrophilic) Daily Wear Contact Lenses) 脊柱钢板系统(Spinal Plating Systems) 手术缝合线(Surgical Sutures)
FDA 将继续发布指南,将此“基于安全和性能的路径”(Safety and Performance Based Pathway)应用于其他更多 FDA 认同的相应性能标准的器械类型。业界(Industry)可以就 FDA 应考虑认同哪些类型的器械性能标准(Performance criteria)提出建议。例如,业界可以就 FDA 认同(FDA-recognized)的、具有全面共识标准的器械提出建议。据 FDA 介绍,FDA 鼓励行业及其他利益相关方就符合条件的器械类型应采用何种性能标准提交基于证据的建议。
如何通过基于安全和性能的路径准备 510(k)(How to prepare a 510(k) using the Safety and Performance Based Pathway)
根据基于安全和性能的路径,支持实质等同 SE 认定所需的信息量和类型将取决于性能标准和测试方法的来源。FDA 建议申报方在其 510(k) 申报的封面函(Cover letter )中明确声明其提交旨在通过基于安全和性能的路径进行审核。
重要的是,510(k) 申报方仍然需要确定实质等同 SE 某些方面的对比器械。但制造商可以选择在适当的情况下,通过此路径证明实质等同 SE,而无需通过直接比较测试(Comparison testing)来证明器械与对比器械具有同等的安全性和有效性。
对医疗器械的修改是否需要新的 510k(Is a new 510(k) required for a modification to the device)
介绍(Introduction)
由于多种因素,医疗器械的设计和材料(Design and materials)会频繁修改;例如供应链的变化、持续的工艺改进,或为了跟上能够改善医疗器械在临床环境中运作效能的技术创新。对器械的重大修改(Major modifications)可能需要 FDA 进行上市前审查(Premarket review),而微小修改则可能无需进行。
当受 510(k) 要求约束的合法上市医疗器械在设计(Design)、组件(Components)、制造方法(Method of manufacture)或预期用途方面(Intended use)发生重大变更或修改时(Significantly changed or modified),必须提交上市前通知 510(k)。重大变更或修改是指可能显著影响器械安全性或有效性的变更或修改,或器械预期用途的重大变更或修改(21 CFR 807.81(a)(3))。
监管条款(Regulatory language)提供了必要的灵活性,而非僵化地限定需要经 FDA 审查的变更类型。恰当运用这种灵活性有助于促进创新,并确保 FDA 仅在必要时介入进行监管。对于某些类型的器械变更,FDA 认为无需提交新的 510(k),依赖现有的质量体系 QS(Quality System)要求(21 CFR 820)是合理确保变更器械安全性和有效性的最简便方法(The least burdensome approach)。
FDA 发布了以下两份指南,旨在帮助相关方确定合法上市器械的变更何时需要提交新的上市前通知 510(k)。这些指南旨在明确哪些变更会触发 FDA 审查要求,并通过提供最少负担的方案(Least burdensome approach),详细阐述此类决策背后所依据的监管框架、政策和实践,增强“何时提交”(When to submit)决策过程的可预测性、一致性和透明度。
何时提交现有器械变更的 510(k)(一般修改指南):https://www.fda.gov/media/99812/download 何时提交现有器械软件变更的 510(k)(软件修改指南):https://www.fda.gov/media/99785/download
这些指南清晰地定义了与器械修改相关的关键术语,并阐述了制造商应如何运用基于风险的评估(例如 ISO 14971)来评估变更是否可能需要提交新的 510(k)。指南还包含流程图、大量示例说明以及关于正确记录器械变更的建议。
在使用这些指南时,制造商必须牢记这些指南中概述的指导原则。
可能需要提交新的 510(k) 的修改示例包括但不限于以下情况:
适应症从处方药改为非处方药 增加了新的患者群体 使用环境的变更,例如从专业用途转为家庭用途,或从医院用途(Hospital use)转为门诊运输(Ambulatory transport) 使用频率或使用时长发生变化 更改以表明与某类器械、组件或配件的兼容性(Compatibility),该类器械、组件或配件此前未被标注为与已获准器械兼容 灭菌、清洁或消毒方法的变更 包装完整性或保质期声明的变更 器械设计的变更(Device design) 采用无线通信的变更 患者人为因素或用户界面的变更 材料类型、配方或化学成分的变更 体外诊断器械 VID 中抗体、检测试剂、关键反应组分或结合物的变更
软件特定的变更(Software Specific Changes)
FDA 发布了单独的软件变更指南, 以处理软件特有的变更事项。该指南适用于受 510(k) 要求约束的合法上市医疗器械的软件变更。《软件修改指南》(Software Modifications guidance)旨在补充《一般修改指南》(General Modifications guidance),并专门针对快速的软件开发周期量身定制通用政策(General policies),并更加侧重于风险评估。
软件修改(Software modifications)可能有多种名称,例如:错误修复(Bug fix)、热补丁(Hot patch)、软件变更(Software change)或调整(Tweak)。无论制造商如何分类此类变更,根据质量体系法规(QS regulation)(21 CFR Part 820),这些均被视为设计变更(Design changes)。
所有非软件变更(Non-software changes),即使是对包含软件的医疗器械进行的变更,均受《一般修改指南》(General Modifications guidance)的约束。如果除了软件变更外,还存在影响标签或硬件的多项变更,制造商应同时使用一般修改指南和软件专用修改指南来评估这些变更。如果使用其中任何一项指南得出“新的 510(k)”(New 510(k))结论,则可能需要提交新的 510(k)。
可能需要提交新的 510(k) 的软件修改示例包括但不限于以下情况:
可能引入新风险或改变现有风险的变更,且该变更有可能导致重大损害(Significant harm) 为防止重大危害而对风险控制措施进行的变更 对器械临床功能或性能规格产生重大影响(Significantly affect)的变更
针对现有医疗器械的变更提交新的 510(k)(Submission of a New 510(k) for a Change to an Existing Device)
没有关于对现有 510(k) 进行 510(k) 修改或补充的规定(Provisions)。如果确定修改不包含在当前 510(k) 中,则必须提交新的 510(k)。
如果修改(Modification)不影响器械的预期用途或改变其基本科学技术(Fundamental scientific technology),则特殊 510(k) 可能是合适的。在特殊 510(k) 申报(Special 510(k))中,设计控制流程(Design control process)生成的摘要信息(Summary information)可作为批准提交的依据(Basis for clearing the submission),同时需满足 21 CFR 807.87 中规定的 510(k) 提交所需要素要求。
以上便是关于美国进口清关流程中 FDA 监管产品医疗器械上市前通知(Premarket Notification)510(k) 和 510(k) 提交类型的介绍。












